
Abstract
KRAS mutations have been detected in approximately 30% of all human tumors, and have been shown to predict response to some targeted therapies. The most common KRAS mutation-detection strategy consists of conventional PCR and direct sequencing. This approach has a 10–20% detection sensitivity depending on whether pyrosequencing or Sanger sequencing is used. To improve detection sensitivity, we compared our conventional method with the recently described co-amplification-at-lower denaturation-temperature PCR (COLD-PCR) method, which selectively amplifies minority alleles. In COLD-PCR, the critical denaturation temperature is lowered to 80°C (vs 94°C in conventional PCR). The sensitivity of COLD-PCR was determined by assessing serial dilutions. Fifty clinical samples were used, including 20 fresh bone-marrow aspirate specimens and the formalin-fixed paraffin-embedded (FFPE) tissue of 30 solid tumors. Implementation of COLD-PCR was straightforward and required no additional cost for reagents or instruments. The method was specific and reproducible. COLD-PCR successfully detected mutations in all samples that were positive by conventional PCR, and enhanced the mutant-to-wild-type ratio by >4.74-fold, increasing the mutation detection sensitivity to 1.5%. The enhancement of mutation detection by COLD-PCR inversely correlated with the tumor-cell percentage in a sample. In conclusion, we validated the utility and superior sensitivity of COLD-PCR for detecting KRAS mutations in a variety of hematopoietic and solid tumors using either fresh or fixed, paraffin-embedded tissue.
Similar content being viewed by others

Main
KRAS, a member of the RAS oncogene family, plays a key role in RAS/MAPK signaling, which is involved in multiple cellular processes, including proliferation, differentiation and apoptosis.1 Activating mutations of the KRAS gene impair GTPase activity resulting in constitutive activation. Up to 30% of all human tumors harbor an activating mutation in this gene, most commonly in lung, colon, and pancreatic cancers, and hematopoietic neoplasms.2 Screening tumors for possible KRAS mutation has multiple clinical applications and is gaining increasing attention in clinical practice. For instance, in lymphoma patients with KRAS mutations quantitative assessment of mutation levels in follow-up specimens provides an effective way to monitor and quantify residual disease and to detect relapse.3 In lung cancer patients, KRAS mutation is an adverse prognostic factor.4, 5 Accumulating evidence has shown that KRAS mutation status can be highly predictive for patient response to anti-epidermal growth factor receptor (EGFR) antibody therapies, such as cetuximab and panitumumab, in colorectal cancer, lung, and head and neck cancers.6, 7, 8, 9 These therapies are exclusively effective in tumors with wild-type KRAS.10, 11, 12, 13 In colorectal cancer patients, in particular, KRAS mutation testing has quickly become standard-of-care.14 In the era of targeted therapy and personalized medicine, requests for KRAS mutation testing will increase, and are likely to do so exponentially. In the molecular diagnostic laboratory at our institution, a doubling of requests for KRAS mutation occurred in the year 2008.
Currently, the gold standard for KRAS mutation detection is conventional PCR amplification followed by direct sequencing.15 Most other detection methods reported in literature16, 17, 18, 19 also involve PCR. PCR amplification enables mutation detection from a very small amount of samples. However, conventional PCR methods do not selectively amplify the mutant sequences in tumor specimens, which is needed for optimal sensitivity in specimens with a low tumor-cell count or high non-neoplastic cell content. On the contrary, in conventional PCR the predominant wild-type sequences have a competitive advantage over mutant sequences in being amplified, leaving the mutant sequences more difficult to be detected by downstream methods. To improve the sensitivity of mutation detection, tumor-cell enrichment methods, such as cell sorting or microdissection, are commonly required before DNA extraction for PCR. However, tumor-cell enrichment is not always achievable, particularly in samples with abundant desmoplastic stroma surrounding a minority of tumor cells and abundant inflammatory cells, or in scanty tumor cells after chemotherapy or radiation therapy in the follow-up setting.20, 21 As a result, conventional PCR-based assays are limited in their ability to identify low levels of mutation-bearing tumor cells and this may greatly effect on therapeutic decisions.
Co-amplification-at-lower denaturation-temperature PCR (COLD-PCR) is a novel modification of the conventional PCR method that selectively amplifies minority alleles from a mixture of wild-type and mutant sequences irrespective of the mutation type or position within the sequence.22 This method is based on the observation that there is a critical denaturation temperature (Tc) for each DNA sequence, which is lower than its melting temperature (Tm). PCR amplification efficiency for a DNA sequence drops abruptly if the denaturation temperature is set below its Tc.
In this study, we compared COLD-PCR with the conventional PCR for the detection of KRAS mutations in 50 clinical samples. The data show that COLD-PCR is up to four times more sensitive than the conventional PCR method, able to detect 1.5% of tumor cells with KRAS mutation. COLD-PCR was also equal to the conventional PCR method in its reproducibility and did not require additional costs in either reagents or instruments.
Materials and methods
Clinical Samples and Genomic DNA Extraction
Clinical specimens were obtained from archival DNA in the Clinical Molecular Diagnostics Laboratory at The University of Texas MD Anderson Cancer Center. A total of 50 samples formed the study group, including DNA extracted from 20 fresh bone-marrow aspirate specimens involving hematopoietic neoplasms, and DNA extracted from 30 solid tumor specimens that were routinely fixed in formalin and paraffin embedded. The clinical characteristics of the patients from whom these specimens were derived are listed in Table 1. All samples had been tested earlier for KRAS codon 12 and 13 mutations as part of their clinical work up using our routine pyrosequencing analysis following conventional PCR. Genomic DNA from fresh bone-marrow aspirate specimens was extracted using the Gentra AUTOPURE LSTM automated DNA extraction system (Qiagen Systems, Valencia, CA, USA). The percentage of tumor cells in each specimen was obtained from a review of the manual differential counts listed in the pathology reports associated with each case. For solid tumor specimens, the hematoxylin–eosin stained slides were reviewed, and the percentage of tumor was estimated as a percentage of tumor cells to non-neoplastic cells by visual inspection. Using the hematoxylin–eosin-stained slide as a guide, tumor areas on the unstained formalin-fixed paraffin-embedded (FFPE) sections were manually micro-dissected and DNA was extracted using the QIAmp DNA mini kit (Qiagen Systems).
Detection of KRAS Mutation by Conventional PCR and Pyrosequencing
KRAS mutation testing has been routinely carried out in our laboratory using conventional PCR followed by pyrosequencing. An amplicon of 98 bp is amplified using a forward primer, 5′-TATAAACTTGTGGTAGTTGG-3′, and a reverse biotinylated primer, 5′-biotin-ATTGTTGGATCATATTCGT-3′. The PCR master mix contains the forward and reverse primers (each 0.2 μmol/l), 250 μmol/l of dNTP mix, 2.5 mmol/l MgCl2, 1 × PCR buffer (Applied Biosystems, Carlsbad, CA, USA), 1 U of AmpliTaq Gold and 200 ng of sample genomic DNA in a total volume of 50 μl. PCR-cycling conditions consist of initial denaturing at 95°C for 2 min; 49 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s; and final extension at 72°C for 10 min. The reactions are carried out on an ABI 2720 Thermocycler (Applied Biosystems). All samples are run in duplicate and patient samples are processed along with a positive control sample (positive for the presence of a GGT-to-GCT mutation at codon 12), a negative control sample and a reagent control. The PCR products are electrophoresed in an agarose gel to confirm successful amplification of the 98-bp PCR product before pyrosequencing. The PCR products (each 15 μl) are then sequenced by the Pyrosequencing PSQ96 HS System (Biotage AB, Uppsala, Sweden), following the manufacturer's instructions using the pyrosequencing primer, 5′-CTTGTGGTAGTTGGAGCT-3′. Each sample is sequenced with two separate programs of nucleotide dispensation orders: 5′-TACGACTGC-3′, designed for detecting mutations at codon 12; and 5′-TGTATCGATCGT-3′, designed for detecting mutations at codon 13. This primer and the dispensation orders enable us to capture all possible mutations of the wild-type sequence of GGT GGC at codons 12 and 13 of KRAS.
Cold-PCR for KRAS Mutation Detection
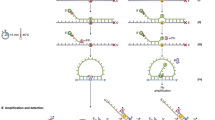
We applied the COLD-PCR assay for detecting KRAS mutations based on the methodology described in the literature,22 with several modifications. A key consideration in designing a COLD-PCR assay that selectively amplifies the minority mutant alleles is to determine a new reduced denaturation temperature for the reaction. Ideally, this reduced denaturation temperature allows mainly the heteroduplexes to be denatured and amplified, and leaves the homoduplexes double-stranded and not amplified efficiently. We used the same primers as used in our conventional PCR assay, which produces an amplicon of 98 bp with 41.8% of GC content and a Tm of 70.9°C. Using the Poland algorithm,23 we plotted Tm profiles of the wild-type sequence along with those of mismatched sequences at each base pair within KRAS codons 12 and 13 (Figure 1). On the basis of this information, we set the reduced denaturation temperature of the COLD-PCR reaction at 80°C. The reaction protocol started with 10 cycles of the conventional PCR amplification for an initial buildup of all amplicons, followed by 40 COLD-PCR cycles to selectively enrich for mutant sequences. The initial conventional PCR cycling conditions are summarized as follows: 95°C for 10 min; 10 cycles at 95°C for 15 s, 57°C for 30 s, and 72°C for 1 min; 72°C for 7 min; and 95°C for 2 min. Then 40 cycles of COLD-PCR were performed at 95°C for 15 s, 70°C for 8 min, 80°C for 3 s, 55°C for 30 s, and 72°C for 1 min (Figure 1). Full COLD-PCR reactions were performed on an ABI 2720 Thermocycler (Applied Biosystems). The COLD-PCR products were electrophoresed in agarose gels to confirm successful amplification of the 98-bp PCR product prior to pyrosequencing as described above. As with our routine sequencing assay, a positive control, a negative control and a reagent control were included in each run. All samples were run in duplicate.

Design of the full co-amplification at lower denaturation temperature PCR COLD-PCR assay for KRAS mutations detection. (a) Nucleotide sequence of the 98-bp PCR product and melting temperature (Tm) profiles of the wild-type and mutant sequences at each of the six nucleotides of codons 12–13. Arrows indicate primer sequences (underlined). Sequence of codons 12–13 is indicated in red. Figure legends indicate Tm profiles for wild-type (‘wild’) and mutant (‘m’ and nucleotide number, eg ‘m25’) sequences. (b) Description of the PCR reaction conditions used in this study for conventional PCR and COLD-PCR protocols.
Comparison of Sensitivities of Cold-PCR and Conventional PCR
We performed a serial dilution study to examine the enhancement of the sensitivity of mutation detection by COLD-PCR in comparison with conventional PCR. A patient DNA sample containing a GGT to GCT mutation at codon 12 was used as the source of the mutant allele. This mutation-containing DNA sample was serially diluted with a wild-type DNA sample to obtain 1:2, 1:4, 1:8, 1:16, 1:32, 1:40 and 1:50 mutant-to-wild-type mixtures. All of the DNA mixtures were simultaneously subjected to conventional PCR and COLD-PCR followed by pyrosequencing, as described above. All samples were run in duplicate. The mutant-to-wild-type ratio was defined as the ratio of the peak height of a single-mutant nucleotide over the peak height of a single wild-type nucleotide on pyrograms. The enhancements were represented as fold changes of the average mutation-to-wild-type ratios of the same dilution between COLD-PCR and conventional PCR amplifications.
Statistical Analysis
Student t-tests were carried out wherever applicable. A P-value of <0.05 was considered statistically significant. Associations between two variables were determined by calculating the Pearson correlation coefficient.
Results
Determination of the Sensitivity with Dilution Study
Figure 2a shows the pyrograms from the dilution study. With the conventional PCR amplification, the mutant-nucleotide peak on the pyrogram became indistinguishable at 1:8 dilution, whereas it was clearly present at 1:32 dilution with COLD-PCR. As the mutant sample we used for the dilution study was heterozygous at KRAS codon 12, a 1:32 dilution translates to approximately 1.5% of the detection sensitivity, which is four times better than the 6% sensitivity obtained with the conventional PCR method.

Sensitivity determined by dilution study. (a) Representative pyrograms of serial-dilutions study with conventional PCR and co-amplification-at-lower denaturation-temperature PCR (COLD-PCR). (b) Comparison of pyrosequencing results from undiluted to1:32 dilution after conventional PCR and COLD-PCR (bars), and enhancement of COLD-PCR in serial dilutions (line).
As shown in Figure 2b, with increasing serial dilutions, the mutation-to-wild-type ratio decreased proportionally in a first-order kinetics manner in both conventional PCR and COLD-PCR. The enhancement of mutation detection by COLD-PCR increased exponentially with increasing dilution. With the original specimen, which contains approximately 50% mutant allele, COLD-PCR and conventional PCR had roughly the same efficiency in amplifying mutant alleles. At 1:32 dilution, COLD-PCR showed a sixfold greater efficiency in amplifying mutant allele compared with conventional PCR.
Within the detectable dilution range, all duplicate analyses of COLD-PCR products produced consistent pyrosequencing results. Different runs from the same sample also generated very similar results (correlation coefficient, 0.923).
Comparison of KRAS Mutation Detection Using Conventional PCR and COLD-PCR in Patient Samples
With the sensitivity of the COLD-PCR method obtained from the dilution study above, we further examined the potential enhancement of KRAS mutation detection by COLD-PCR over conventional PCR in different types of clinical samples. A total of 50 patient samples were tested, including 20 fresh bone-marrow aspirate samples from patients with acute leukemia (mostly myeloid with one lymphoblastic) and myelodysplastic syndromes (MDS) and 30 fixed paraffin-embedded samples of adenocarcinomas arising in the colon, rectum, liver, lung, bladder and pancreas (primary neoplasms or sites of metastasis; Table 1). The results using conventional PCR and COLD-PCR are summarized in Tables 2 and 3. In all 35 samples were shown to harbor KRAS mutations at codon 12, 2 had mutations of codon 13, and 13 were negative for mutation.
In most cases, COLD-PCR enhanced KRAS mutation detection by up to 4.74-fold, with an average enhancement of 1.55-fold ±0.76. Moreover, in a patient with acute myeloid leukemia after chemotherapy, in a follow-up bone marrow sample that tested negative for KRAS mutations by conventional PCR, COLD-PCR showed a GTT–to-GAT mutation at codon 12 (Figure 3b). This same mutation was present at the time of an initial diagnosis, three months earlier.

Comparison of pyrograms of sample no.48 after conventional PCR and co-amplification-at-lower denaturation-temperature PCR (COLD-PCR) amplification. The arrow indicates a G12D mutation identified by COLD-PCR, which is not visible in conventional PCR.
Mutation detection in fixed, paraffin-embedded specimens of solid tumors (1.67±0.95) was also enhanced by COLD-PCR. In particular, COLD-PCR showed significant higher enhancement in the solid tumor samples, in which the mutant-to-wild-type ratios by conventional PCR were less than 50% (1.94±0.93 vs 0.86±0.36, P=0.022). Neither location or type of solid tumors nor the type of hematopoietic neoplasms had any effect on enhancement. The tumor-cell count or percentage did not correlate with the mutant-to-wild-type peak ratio in pyrograms using either conventional PCR or COLD-PCR (correlation coefficient of 0.452 and 0.244, respectively). However, lower tumor-cell content in the specimens was significantly associated with greater enhancement; the average enhancement for samples with 50% or less tumor cells was 1.89±0.88, vs 1.34±0.26 in samples with higher than 50% tumor cells (P=0.012). In four cases in which COLD-PCR failed to improve the mutation detection (enhancement <1), the tumor cell percentages were over 80%.
A total of eight different types of KRAS point mutations at codons 12 or 13 were detected in these samples by pyrosequencing (Table 2). All but two of these mutations were at codon 12. These mutations involved four G–A transitions (G12D, G12S, G13D, G13S), two G–T transversions (G12V, G12C) and two G–C transversions (G12A, G12R). The frequency of each type of mutation at codon 12 (listed in decreasing order) and its percentage in all positive samples from this study were: G12D (GAT, 49%) >G12V (GTT, 24%) >G12R (CGT, 11%) >G12S (AGT, 5%) >G12A (GCT, 3%) or G12C (TGT, 3%). The G12D mutation was most frequent in acute myeloid leukemias (seven of nine positive cases), whereas the G12D and G12V mutations were most frequent in colorectal carcinoma (nine and seven in 19 positive cases, respectively). The two cases with mutations at codon 13 were a case of MDS (G13S) and a case of colon carcinoma (G13D). Enhancement by COLD-PCR among different mutation types (nucleotide exchanges and locations) did not show any significant differences.
Discussion
The main purpose of this study was to evaluate the potential of COLD-PCR to enhance the detection of KRAS mutations in clinical samples. The current sensitivity of the KRAS mutation test in our laboratory using conventional PCR methods followed by pyrosequencing is approximately 10%. This requires that specimens submitted for testing have approximately 20% tumor cells, accounting for the fact that each neoplastic cell may be heterozygous and thus may carry only one mutated allele. In our laboratory, solid tumor samples are routinely micro-dissected to enrich tumor-cell content, but this approach can be challenging when using small biopsy specimens. Enhancing the sensitivity of the KRAS assay potentially eliminates this tumor-cell enrichment step, saving manpower and time.
Our dilution study demonstrated that COLD-PCR increased the KRAS mutation detection sensitivity from 1:8 to 1:32 (fourfold), and that this approach is both reliable and reproducible. Adoption of the method was straightforward, requiring no additional cost for reagents and instruments. With its selective enhancement of minority mutant-allele amplification, the resultant increased sensitivity also could potentially shorten the overall turnaround time by reducing the repeats needed when results are equivocal using conventional PCR methods.
In this study, we identified a total of eight different point mutations, six at codon 12 and two at codon 13. Mutations at codon 12 were by far the most common, found in 35 of the 37 mutation-positive samples. There was roughly an equal frequency of transition and transversion mutations identified in these samples, even though transition mutations have been found to be about an order of magnitude more common than transversions among all point mutations.24 Mutations that switch nucleotide types may also slightly affect the melting temperature,25, 26 which might change the Tc of the COLD-PCR reaction. However, using a fixed Tc for all types of mutations in this study, we did not observe a significant difference in the enhancements by COLD-PCR among mutation types, suggesting that this effect may be insignificant.
As the dilution study results indicated, enhancement by COLD-PCR was non-linear and more prominent when the concentrations of mutation-containing sequences were lower. The most likely explanation is that during each cycle of COLD-PCR, only the heteroduplexes are denatured and subsequently amplified. In an ideal situation, with sufficient amplification cycles, the amplification product at the end of the reaction will contain almost equal amounts of mutant and wild-type sequences. Therefore, the maximum mutant-to-wild-type ratio in COLD-PCR products should be close to 1.
The lower enhancement by COLD-PCR observed in our study (<2-fold on average) as compared with the 3–12-fold enhancement reported earlier by others may be explained by decisions we routinely make before analysis.22 First, we require all submitted samples to contain at least 20% tumor cells, and we routinely carry out micro-dissection on fixed, paraffin-embedded samples to enrich tumor-cell content. Second, our conventional PCR reactions comprise 50 cycles compared with 40 cycles in a study reported earlier. Both of these steps boost the concentration of mutation-containing sequences, and therefore minimize the potential for enhancement of mutation by COLD-PCR to further enrich the mutant sequence. In addition, a whole genome amplification (WGA) step was applied to all the specimens before COLD-PCR in the reported study. It is also possible that WGA further dilutes the mutant allele concentrations.
An intriguing issue with increasing the sensitivity of KRAS mutation detection is understanding what level of mutation has clinical significance. Will the ability to detect very low levels of mutation, on the order of 1–2%, be as significant as a high level? Although it has been shown that patients with carcinomas that have KRAS mutations as well as patients with KRAS wild-type tumors do not benefit from anti-EGFR therapy, one may argue that patients with a low level of KRAS mutations could still benefit from the therapy. Clinical trials are necessary to assess the quantitative effects of KRAS mutation on patient response to EGFR antibody therapy.
In conclusion, our findings confirm that the COLD-PCR method can increase the sensitivity of KRAS mutation detection in various clinical samples, including DNA extracted from either fresh or fixed, paraffin-embedded tissue specimens. COLD-PCR can be a good strategy for mutation detection in specimens with high non-neoplastic cell content, in small specimens in which neoplastic cells are difficult to micro-dissect and therefore enrich, and whenever a mutation is suspected to be present but is undetectable using conventional PCR and sequencing methods.
References
Karnoub AE, Weinberg RA . Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008;9:517–531.
van der Weyden L, Adams DJ . The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta 2007;1776:58–85.
Cikota BM, Tukic LJ, Tarabar OT, et al. Detection of t(14;18), P53 and RAS gene mutations and quantification of residual disease in patients with B-cell non-Hodgkin's lymphoma. J Exp Clin Cancer Res 2007;26:535–542.
Slebos RJ, Kibbelaar RE, Dalesio O, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med 1990;323:561–565.
Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005;23:5900–5909.
Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res 2007;13:2890–2896.
De Roock W, Piessevaux H, De Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 2008;19:508–515.
Di Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 2007;96:1166–1169.
Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992–3995.
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626–1634.
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–2648.
Di Fiore F, Charbonnier F, Lefebure B, et al. Clinical interest of KRAS mutation detection in blood for anti-EGFR therapies in metastatic colorectal cancer. Br J Cancer 2008;99:551–552.
Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25:3230–3237.
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–1765.
Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by pyrosequencing. J Mol Diagn 2005;7:413–421.
Amicarelli G, Shehi E, Makrigiorgos GM, et al. FLAG assay as a novel method for real-time signal generation during PCR: application to detection and genotyping of KRAS codon 12 mutations. Nucleic Acids Res 2007;35:e131.
Do H, Krypuy M, Mitchell PL, et al. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. BMC Cancer 2008;8:142.
Hayes VM, Westra JL, Verlind E, et al. New comprehensive denaturing-gradient-gel- electrophoresis assay for KRAS mutation detection applied to paraffin-embedded tumours. Genes Chromosomes Cancer 2000;29:309–314.
Shi C, Fukushima N, Abe T, et al. Sensitive and quantitative detection of KRAS2 gene mutations in pancreatic duct juice differentiates patients with pancreatic cancer from chronic pancreatitis, potential for early detection. Cancer Biol Ther 2008;7:353–360.
Fischer C, Buthe J, Nollau P, et al. Enrichment of mutant KRAS alleles in pancreatic juice by subtractive iterative polymerase chain reaction. Lab Invest 2001;81:827–831.
Li J, Berbeco R, Distel RJ, et al. s-RT-MELT for rapid mutation scanning using enzymatic selection and real time DNA-melting: new potential for multiplex genetic analysis. Nucleic Acids Res 2007;35:e84.
Li J, Wang L, Mamon H, et al. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nature Medicine 2008;14:579–584.
Steger G . Thermal denaturation of double-stranded nucleic acids: prediction of temperatures critical for gradient gel electrophoresis and polymerase chain reaction. Nucleic Acids Res 1994;22:2760–2768.
Drake JW . Chaos and order in spontaneous mutation. Genetics 2006;173:1–8.
Lankas F, Sponer J, Langowski J, et al. DNA basepair step deformability inferred from molecular dynamics simulations. Biophys J 2003;85:2872–2883.
Naiser T, Ehler O, Kayser J, et al. Impact of point-mutations on the hybridization affinity of surface-bound DNA/DNA and RNA/DNA oligonucleotide-duplexes: comparison of single base mismatches and base bulges. BMC Biotechnol 2008;8:48.
Acknowledgements
The authors wish to thank James Gilbert for his excellent support in preparing the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Zuo, Z., Chen, S., Chandra, P. et al. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol 22, 1023–1031 (2009). https://doi.org/10.1038/modpathol.2009.59
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2009.59
Keywords
This article is cited by
-
Limitations and opportunities of technologies for the analysis of cell-free DNA in cancer diagnostics
Nature Biomedical Engineering (2022)
-
Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer
Nature Reviews Gastroenterology & Hepatology (2020)
-
A new primer construction technique that effectively increases amplification of rare mutant templates in samples
BMC Biotechnology (2019)
-
Next generation sequencing of exceptional responders with BRAF-mutant melanoma: implications for sensitivity and resistance
BMC Cancer (2015)
-
A phase 1 study of intermittently administered pazopanib in combination with continuous daily dosing of lapatinib in patients with solid tumors
Cancer Chemotherapy and Pharmacology (2015)
