
Abstract
Though not required currently for staging, regression is a histopathologic parameter typically reported upon diagnosis of an invasive primary cutaneous melanoma. The studies examining the prognostic significance of regression in patient outcome have yielded controversial findings; likely because the definition and assessment of regression have not been consistent, in addition to subjectivity of pathologists’ interpretation. Regression is histologically characterized by variable decrease in the number of melanoma cells accompanied by the presence of a host response consisting of dermal fibrosis, inflammatory infiltrate, melanophages, ectatic blood vessels, epidermal attenuation, and/or apoptosis of keratinocytes or melanocytes; the relative extent of these features depends on the stage of the regression. However, the magnitudes to which these individual changes must be present to meet the threshold of histologic regression have not been well defined or agreed upon, and thus, the definition and classification of histologic regression in melanoma varies considerably among institutions and even among individual pathologists. In order to determine the clinical significance of histologic analysis of regression, there is a compelling need for a universal scheme to objectively define and assess histologic regression in primary cutaneous melanoma, so that the biologic and prognostic significance of this process may be completely understood.
Similar content being viewed by others

Introduction
Primary cutaneous melanoma accounts for approximately 3% of all malignant skin tumors but contributes to most skin cancer–related deaths.1 There are treatment options for most melanomas, primarily based on the stage of the disease, including surgery (wide local excision with or without sentinel lymph node (SLN) biopsy or regional lymph node dissection), adjuvant or neo-adjuvant therapy (chemotherapy, radiation, targeted therapy, immunotherapy). Breslow thickness is the most important histologic parameter predicting outcome in primary cutaneous melanomas. Lesions with thickness ≤1.00 mm usually have a very good prognosis; more than 95% of patients with such ‘thin’ melanomas are alive at 10 years after diagnosis. However, some patients with a thin lesion develop metastasis, and thus, it is controversial whether sentinel lymphadenectomy should be performed in addition to wide local excision for primary lesions ≤1.00 mm. Furthermore, it is unclear if other prognostic factors may help reduce the number of patients receiving SLN biopsy in lesions thicker ≥1 mm. The 2016 recommendations of the American Joint Committee on Cancer (AJCC) for thin melanomas will include ulceration or tumor thickness >0.8 mm as features supporting recommendation of SLN biopsy (stage pT1b). And, although it is not included in the AJCC recommendation, in some institutions, histologic regression also is considered to be an indicator for SLN biopsy in patients with a thin melanoma.2, 3, 4
From an immune point of view, melanoma, like other solid tumors, may regress spontaneously with partial or complete disappearance of tumor.5 Regression is a relatively common event in melanoma,6 the overall incidence ranging from 10 to 35%,7 and has been reported in up to 58% of thin melanomas (Breslow thickness ≤1.00 mm). It is likely that the observed differences in regression rates are primarily due to inconsistencies in the definition and assessment of regression among the studies, highlighting the need for more defined histologic criteria. Regression may be an important prognostic indicator, since several cases of melanoma presenting initially as regional metastases (eg, cutaneous or lymph node) have been documented in the setting of complete regression of the primary melanoma or an unknown primary tumor,8, 9, 10, 11, 12 suggesting an association between regression and metastasis. In contrast, other more recent studies have indicated that the presence of histologic regression in primary melanoma is associated with negative SLN status.13 Thus, the prognostic impact of regression in patients with melanoma remains controversial. In this review, we discuss the various definitions and measurements of melanoma regression, the potential value of regression as a prognostic parameter, and the possible biologic mechanisms contributing to regression with the goal to promote the need for more standardized criteria for histologic assessment of regression.
Definition and classification of regression in primary melanoma
Regression of malignant tumors can occur spontaneously or in response to treatment. Spontaneous regression is defined as partial or complete disappearance of a previously documented tumor in the absence of therapeutic interventions or physical trauma.14 Regression of primary melanoma is characterized clinically by initial (subtle and often unnoticed) hyperpigmentation, followed by depigmentation of part of or the entire lesion, resulting in blue, pink, white, or gray areas.
There is no still standard definition for histologic regression of primary melanoma. In general, however, most authors define histologic regression of primary melanoma as partial, segmental or complete replacement of melanoma cells with a variable host response. This host response includes variably dense mononuclear infiltrate, melanophages, and/or dermal fibrosis accompanied by increased dermal vascularity, with variable epidermal attenuation.15, 16, 17 Notably, histologic regression is often seen in the absence of clinically documented regression.
Mechanisms of regression in melanocytic lesions
The etiology of regression in melanocytic lesions is multifactorial, and the contributing mechanisms are yet to be completely elucidated.18 Our current understanding indicates that host-immune-mediated responses, particularly through CD8-positive cytotoxic T lymphocytes (CTLs) play a significant role in the development of regression.19 The role of the immune system in regression is further supported by the high incidence of CD4-positive T lymphocytes and Th1 cytokines in regressed tumors,18 as well as the high levels of tumor-specific antibodies and CTLs in the peripheral blood of patients with regressed tumors.19
Some authors have suggested that regression may be the biologic effect of the inflammatory response,20 directed against specific melanocyte-associated antigens such as melan-A (also known as MART-1).21 Moreover, a dense lymphocytic infiltrate, a feature of early regression also seen in halo nevi, lends support to the key role of lymphocytes in mediating regression. This immune-mediated destruction of melanoma cells is also supported by the increasing efficacy of immunomodulatory therapy in melanomas, which induce or enhance antitumor host-immune response.22, 23 However, strictly speaking, just the presence of a dense inflammatory infiltrate with no evidence of at least partial loss of melanocytes, that is, tumor-infiltrating lymphocytes (TILs) does not fulfill the histologic criteria for regression.24, 25
T lymphocytes
T lymphocytes play a role in the regression of human tumors by recognizing specific antigens. The peptides of tumor cell proteins, also known as tumor-rejection antigens, are presented to T cells via the major histocompatibility molecules.26 They are reported to be the targets of antitumor T-cell response because their levels of expression on normal cells are insufficient to be recognized by T lymphocytes.
Six different categories of tumor-rejection antigens have been reported, three of which are involved in regression of melanoma: tumor-specific mutated oncogenes or tumor suppressor genes (cell cycle regulator, cyclin-dependent kinase 4 (CDKN4)), germ cell antigens (normal testicular proteins) and differentiation antigens (enzymes in the melanin synthesis pathway such as tyrosinase).27
Tumor-specific mutated oncogene or tumor suppressor genes
The tumor-specific mutated oncogenes consist of those that encode strictly tumor-specific antigens and are formed mainly through point mutations or gene rearrangements that occur partly during the process of tumorigenesis. Mutations in the CDKN4 gene are not only well-known contributors to melanoma development but also play an important role in spontaneous regression.28 The reported role of point mutations in T-cell response includes support of the (de novo) binding of a peptide to major histocompatibility class I molecules or modification of a peptide that already binds class I molecules, resulting in a new epitope.29
Germ cell antigens
Germ cell antigens are proteins that are normally expressed only in the testis and placenta. In normal tissue their peptides cannot be presented to T lymphocytes because they are not expressed on major histocompatibility molecules. However, these proteins may be expressed at recognizable levels during oncogenesis due to genomic and epigenetic changes inherent to specific tumors and act as tumor-specific antigens.30 In spite of their expression in various tumors, melanoma antigen (MAGE) and cancer-testis antigens are associated primarily with melanoma. Their high level of immunogenicity and tumor specificity make them ideal targets for development of antigen-specific cancer vaccines. In the early 1990s, MAGE-1 was the first tumor antigen that provoked an autologous CTL response in a melanoma patient.31 Since then, MAGE antigens have been used as vaccines in human immunotherapy protocols, resulting in regression of melanomas, as well as other tumor-associated antigens (TAAs), including gp100.32 The second antigen with the capacity to induce both humoral and cellular immune responses is NY-ESO-1 (cancer-testis antigen 1B), which was isolated from an esophageal squamous cell carcinoma.33 Like the MAGEs, NY-ESO-1 is expressed by a wide range of cancers, including melanoma. T cells modified to express NY-ESO-1-specific T-cell receptors have been reported to induce regression in metastatic synovial cell sarcoma and melanoma.34
Differentiation antigens
This category of tumor-rejection antigens comprise differentiation antigens encoded by genes that are expressed only in certain types of tissue. The differentiation antigens of interest in melanoma are proteins involved in melanin synthesis pathways. The gp100 protein, a melanocyte lineage-specific antigen recognized by HMB45 (human melanoma black-45) antibody, is not only an important diagnostic marker of melanoma, but it also plays a role in therapy as well. Owing to its immunogenic epitopes being detected by human CTLs, gp100 has been used extensively in melanoma vaccines.32 Another T-cell receptor that has been used for development of melanoma vaccines is the MART-1-specific T-cell receptor, which was reported to be specific to an HLA-A2-specific MART-1 peptide epitope. Autologous CD8-positive T cells genetically engineered to express MART-1-specific T-cell receptors have demonstrated their effective role in immunotherapy of melanoma by inducing melanoma regression and long-term cures in animal models as well as in human patients.32
Tyrosinase, an oxidase involved in melanin production, is specifically expressed in melanocytes and has been shown to be overexpressed in melanomas. It was also reported to be immunogenic, and tyrosinase-specific CD8-positive T cells can induce lysis of melanoma cell lines. HLA-DR-specific tyrosinase peptide epitopes, which are known to be identified by CD4-positive T cells, have been used in vaccines against melanoma, alone or in combination with other TAAs.32 The immunogenic tyrosinase-related proteins 1 and 2 (TRP-1, TRP-2) are frequently overexpressed in melanoma. TRP-1 is of interest in immunotherapy, and the use of selectively mutated TRP-1 CD8-positive T-cell epitopes is reported to effectively interrupt immunologic tolerance and initiate effective anti-melanoma responses. TRP-2-targeted immune responses have also been reported to be involved in melanoma regression.32
Spontaneous regression in melanoma
Compared to other tumors, melanoma cells particularly express several proteins with strong immunogenic effects, which are considered to be significant targets for major histocompatibility class II and cytotoxic T cells.35 As such, melanomas with high expression of melan-A/MART-1, gp100, or HLA-A-0201 show more frequent spontaneous regression.36, 37 There are several hypotheses about the mechanism of spontaneous regression in primary melanoma and other melanocytic lesions.37, 38 It has been suggested that an immune response could be initiated at the time of development of lymph node metastasis. Up to 30% of melanoma cases with regression develop a concomitant febrile reaction, and approximately 25% of them are associated with cutaneous erysipelas.39 Finally, some melanoma cells may regress in response to vaccines against infectious diseases, including tetanus, diphtheria, and bacillus Calmette-Guérin.40
Multiple research studies over the years since publication of Ehrlich’s tumor immunosurveillance theory have shown that the TAAs could be targets for cancer immunotherapy.41 Although many of these studies concentrated on immune responses against virus-induced tumors, spontaneous tumor regressions were also sporadically observed in human patients, especially after immunization against common pathogens, supporting the role of TAAs of non-viral origin. Bayer-Garner and others have demonstrated that cytotoxic immune responses may be involved in the regression of some tumors. TAAs that have been reported to be targets for CD8-positive T cells include viral antigens, melanocyte differentiation antigens, and cancer-testis antigens.42, 43
Other factors that can induce melanoma regression include decreased microphthalmia transcription factor activity within melanocytes.44 Another mechanism, proposed by Bastian,45 suggests that decreased telomerase activity can lead to abnormal telomere function, and thus induce replicative senescence in melanoma cells, resulting in the generation of apoptotic bodies and a T lymphocyte response.
Murine models in the study of melanoma treatment
Investigations of checkpoint blockade in mouse models and humans have indicated that preclinical transplantable tumor models provide valuable information for clinical application.46 Programmed cell death protein 1 (PD-1)-specific blocking antibodies have been shown to stimulate tumor regression in diverse human malignancies, including melanoma. Coupling PD-1-specific blocking antibodies with other therapeutic options, including tumor vaccines, adoptive T-cell therapy, or other immunoregulatory antibodies, has been shown to enhance their regression-inducing activity in several model systems, helping to guide further development of these therapies.47
The use of in vivo animal models, especially mice, which can simulate true melanoma behavior as well as recapitulate natural tumor progression from in situ to invasion and metastasis, is essential for the following purposes: (1) to fully understand the background mechanisms underlying tumor regression; (2) to develop new drug regimens with better outcome and/or understand resistance/relapse to the currently available treatment options; (3) to better understand the biology of melanoma tumorigenesis; (4) to limit/reduce the expense of the clinical development of immunotherapies; and (5) to prove the safety and therapeutic antitumor activity of new drugs before applying them in clinical trials in humans.46
The mouse is the most widely used preclinical model because of its availability, ease of manipulation, and known genetic background.46 A number of murine melanoma models are used, including xenograft, syngeneic, and genetically engineered models.48, 49 Xenograft models can be easily established by engraftment of cultured human melanoma cells into immune-deficient mice; these models are used mainly to find key oncogenic pathways. Syngeneic xenograft transplantation involves the induction and relocation of melanoma cells in another host of similar type and genetic background. These models are frequently used to assess immunotherapies and interactions between cancer and immune cells, because of the presence of intact immune system, compared with nude mice. In contrast, genetically engineered models use transgenic mice with modified gene expression; these models are often used to determine the mechanisms of melanomagenesis, elucidate the function of specific genes and gene products, and identify optimal targets for therapeutic purposes. Furthermore, they have been used in combination with other tumor induction methods, such as ultraviolet light or chemical carcinogens, to study the risk factors for melanoma development.49
The reported uses of murine models in the validation of potential antitumor therapies include the following: clinical validation of checkpoint blockade (CTL–associated protein 4 (CTLA-4)- or PD-1-specific monoclonal antibodies) in transplantable tumor models; determination of the prognostic importance of intratumoral lymphocytes and CTLA-4-specific monoclonal antibodies, in genetically engineered tumor models; and refinement of chimeric antigen receptor T-cell technology, in humanized mice.50
Immune therapy in melanoma
Two types of immune responses are noted in primary cutaneous melanoma: (1) regression, which is directed against the more superficial component of the lesion and (2) the tumor-infiltrating lymphocyte response, which is directed against the deeper invasive portion of the lesion.51 Although in theory immunomodulatory therapies should be effective in the treatment of primary melanoma, only a proportion of melanomas respond to such treatments. This finding raises the possibility that a subset of melanoma cells expresses stem cell-like molecules, which can selectively display co-stimulatory molecules capable of avoiding host-immune responses or fail to express melanoma-associated differentiation antigens.52, 53, 54
Once melanoma cells metastasize, surgery alone is usually not curative. High-stage patients have a poor prognosis with a median overall survival of 8–10 months and a 5-year survival rate of 10%.55 Different immune therapies have been used for such advanced staged melanomas. Interleukin-2, one of the early immune-mediated therapies, was shown to mediate tumor regression in melanoma and other malignancies.56 Therapy with recombinant high-dose interleukin-2 to induce immune-mediated tumor cell lysis has been widely used in patients with metastatic melanoma with good objective responses in about 16% of treated patients, supporting the idea that immunotherapy can cure some melanomas.57 Approximately half of these patients achieved durable response and long-term survival. However, various life-threatening toxic effects, including vascular leak syndrome, preclude its use and effectiveness in all metastatic melanoma patients.
Interferons (IFN) initially known for their antiviral properties, have been recognized in the recent decades to produce durable antitumor effects in advanced melanoma,58 which are mediated through multiple molecular mechanisms including promotion of Th1-mediated immune response resulting in intratumoral accumulation of CD8-positive T cells and cell-mediated cytotoxicity; enhancement of dendritic cell survival, maturation, and antigen-presenting activity; in addition to antiangiogenic, antiproliferative, pro-apoptotic effects.59, 60, 61 In particular, several IFN-α2 regimens (IFN-α2a, IFN-α2b, IFN-α2c, high-dose IFN-α and Peg-IFN) have been evaluated in the setting of metastatic melanoma and though the response rates are low, up to 15% of patients develop durable or complete response.62 In addition to flu-like side effects in most patients, fewer than 10% of patients develop severe depression and other psychiatric symptoms, which required dose reduction and/or delayed treatment in 50% of these patients.62
The other promising immunotherapy for patients with metastatic melanoma is based on TILs, which are frequently identified in melanoma. Depending in the distribution, the lymphocytic infiltrates are graded as brisk (band-like infiltrate of lymphocytes along the entire invasive border of the melanoma) and non-brisk. To have an antitumor effect, CTLs must have various capabilities, including migrating to the tumor site(s), proliferation, and production of cytokines, thus, inducing tumor cell lysis.63 It was reported that the antitumor activity of CTLs in the tumor microenvironment is less potent and specific than that of TILs grown ex vivo. It has been suggested that these limitations may be due to immune escape mechanisms such as interferon gamma-mediated activation of programmed death-ligand 1 (PD-L1) and increased numbers of regulatory T cells induced by the inflammation caused by CTL infiltrate in the tumor microenvironment.64 Activation of PD-L1 and PD-L2 leads to T-cell dysfunction by engaging with the PD-1 receptor on CTLs.65 Disruption of this interaction and restoration of host CTL antitumor activity against melanoma is the principal immune mechanism behind the new immunotherapy that uses PD-1 and PD-L1 inhibitors.
Cellular immunity is initially activated when T cells recognize expression of intracellular proteins on the surface of antigen-presenting cells bound to specific mixed histocompatibility complex molecules. CTLA-4 expressed on T cells binds to B7.1 and B7.2 on antigen-presenting cells.47 CTLA-4 can inhibit T-cell function via various mechanisms, including inhibition of co-stimulation (caused by binding of CD28 to B7.1 and B7.2) necessary to generate and maintain T-cell activation as well as its expression on regulatory T cells.66 Antibodies to CTLA-4 inhibit the interaction between CTLA-4 and its ligands to restore the function of T cells in the antigen-presenting cells. This is the basis of immune response mediated by ipilimumab, which targets CTLA-4 and was the first immunotherapy to be approved by the U.S. Federal Drug Administration (FDA) for advanced stage melanoma.
CTLA-4 regulates the activation of T cells in early tumor growth, while PD-1 regulates effector T-cell activity in peripheral tissues in response to tumor progression in the later stages of tumor growth.63 CTLA-4, PD-1, its ligands PD-L1 (B7H1) and PD-L2 (B7-DC) and other immune checkpoint molecules are the focus of ongoing studies. The production of an effective immune response is downregulated by binding of PD-1, an inhibitory receptor present on activated T cells, to PD-L1 ligand, which is often present on tumor cells. It was been suggested that antibodies directed against PD-1 (nivolumab, pembrolizumab) or PD-L1 (durvalumab) support tumor regression in patients with advanced melanoma.67 PD-L1 is expressed in various types of tumors, such as melanoma, non-small-cell lung cancer, renal cell carcinoma, gastric cancer, hepatocellular carcinoma, various leukemias, and multiple myeloma.68, 69 It is present in the cytoplasm and plasma membrane of cancer cells. As per current FDA recommendation, expression of PD-1/PD-L1 in ≥1% of melanoma cells may be used as indication for immunotherapy against PD-1/PD-L1.70 Development and clinical application of monoclonal antibodies that inhibit CTLA-4 and PD-1 has been a major milestone in cancer immunotherapy.
Heterogeneity in the definition of histologic regression
Histologic assessment of regression is rife with inconsistencies since there are no universally accepted set of criteria. Many of the studies characterize regression as simply present vs absent, while others have examined it in more detail by including the stage of regression and/or the horizontal extent of involvement.
Some authors have classified regression on the basis of the stage of its evolution into three groups by comparing the relative proportions of fibrosis and inflammatory infiltrate.7, 15, 71 Thus, early stage is characterized by a mononuclear inflammatory cell infiltrate composed predominantly of mature lymphocytes and scattered histiocytes immediately adjacent to melanocytes and keratinocytes and disruption of the dermal and rarely, the junctional melanocytic component (some loss of vertical/radial growth phases). The intermediate stage consists of near complete absence of melanocytes in a portion of the melanoma, along with scattered melanophages, patchy lymphohistiocytic infiltrate, fibroblastic proliferation, mild collagen deposition, and increasing vascularity within superficial dermis. The late stage is characterized by complete absence of melanocytes, well-developed fibrosis involving and expanding the superficial/papillary dermis, vertically oriented delicate and ectatic capillaries, flattened rete ridge pattern, variable numbers of melanophages, and usually a few inflammatory cells.
In 1993, a study from Massachusetts General Hospital modified this classification and categorized histologic regression into early, intermediate, and late stages based on the degree of loss of melanoma cells within the dermis and epidermis in addition to inflammation and fibrosis compared with an adjacent area of the preserved primary melanoma.71 In early regression, the distribution of melanoma cells is disrupted by the dense infiltrates of lymphocytes with minimal degeneration of melanocytes and without obvious fibrosis. In intermediate regression, there is some degeneration of melanoma cells in a background of varying admixtures of lymphoid cells and increased fibrous tissue, variable telangiectasia and melanophages. In late regression, there is prominent loss of melanoma cells while the normal papillary dermis is replaced by horizontally placed collagen fibers, leading to extensive fibrosis with variable telangiectasia, melanophages, sparse lymphocytic infiltrate, and effacement of the epidermis. Some authors have also classified regression as mild, moderate, and severe depending on the extent of involvement of the area of invasive melanoma.72
Botella-Estrada et al2 subdivided regression into two stages: (1) early, characterized by small foci of decreased or absent melanoma cells in the dermal component of the tumor, some degree of dermal fibrosis, and dense inflammatory infiltrate; and (2) late, characterized by a medium to large area of decreased or absence of dermal melanoma cells and very prominent dermal fibrosis. Melanomas have been classified as ‘regressing’ vs ‘regressed’,73 and as ‘active’ vs ‘past’ 74, 75 to reflect similar histopathologic changes. Since the active/early stage of regression overlaps considerably with brisk TILs and is subject to interpretation, many authors document only the presence or absence of late regression (Table 1).
Regression has also been classified according to the area of loss of melanoma within the dermis and epidermis as a percentage of the whole horizontal extent of the melanoma. Based on this estimation, regression is classified into three groups: no or minimal, focal (<50 or <75% of the entire melanoma), and extensive (≥50 or ≥75%).76, 77, 78 While this semiquantitative assessment is typically applied to the entire lesion, in some studies only a representative section of the melanoma has been analyzed for each case.78 Currently, the AJCC classification recommends 75% as the cutoff for distinguishing focal from extensive regression since some studies have identified melanomas with ≥75% regression being associated with metastasis.76, 77
As another attempt to quantify it, regression may be described as partial vs complete. Complete regression of melanoma has traditionally been defined as the absolute absence of any dermal or junctional melanocytes and replacement of the entire lesional area by varying amounts of fibrosis, inflammation, and sometimes abundant melanophages (tumoral melanosis) in the site of a previously documented pigmented lesion.9, 11 In such scenarios, since no melanocytes remain in the lesion, a definitive diagnosis of regressed melanoma cannot be rendered, but the optimal diagnosis would rather be ‘pigment incontinence consistent with a completely regressed melanocytic lesion’. As such, the malignant biologic potential of the regressed lesion is realized only if the patient develops regional or distant metastasis. In contrast, any degree of regression of only a portion or segment of melanoma, with preserved adjacent tumor, is considered to be partial or segmental regression.16 However, some authors differentiate complete from generalized regression as the total absence of melanocytes in epidermis and dermis, even if it is only an area in an otherwise intact melanoma.79
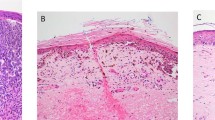
At our institution, we do not routinely report the active/early stage of regression (Figure 1a), due to the considerable overlap between TILs and early stage regression. We define histologic regression as the presence of an area of mononuclear cell infiltrate with associated variable loss of melanoma cells, varying degrees of non-laminated dermal fibrosis, melanophagocytosis, and telangiectatic blood vessels; and variably attenuated epidermis (Figure 1b). This represents the late stage of regression. Such areas may show active degeneration of melanoma cells and/or keratinocytes. We also report the horizontal extent of regression as extensive when >50%. But, following the CAP recommendation, we also indicate when the regression involves >75% of the lesion (Figure 2a–c).

Representative histologic images of different stages of regression of primary cutaneous melanoma. (a) The early/active stage is characterized by a dense dermal lymphocytic infiltrate (black arrowhead) surrounding the melanoma cells (black arrow), some of which may be degenerating (inset), densely and coarsely pigmented macrophages (open white arrow), and minimal dermal fibrosis, if any. (b) The late stage is characterized by dense dermal fibrosis (white arrowhead), numerous melanophages (open white arrow), sparse lymphocytic infiltrate (black arrowhead), scattered dyskeratotic keratinocytes (inset), and almost complete absence of junctional and dermal melanocytes (black arrow) (hematoxylin and eosin stain; magnification: × 100).

Representative histologic images of partial regression of primary cutaneous melanoma classified on the basis of the horizontal extent: (a) focal (<50%), (b) intermediate (>50 to ≤75%), and (c) extensive (>75%) (hematoxylin and eosin stain; magnification: × 100).
Thus, the definition of regression and the histologic classification systems described here are divergent and extremely subjective. The lack of standard classification of regression and the different methods of assessing and documenting regression between different institutions have likely contributed to inconsistent results in the literature. Therefore, a unified set of histologic criteria to assess regression is critical to determine the significance of regression in the outcome of patients with primary cutaneous melanoma.
Prognostic significance of regression in melanoma
Per the College of American Pathologists (CAP) and AJCC, regression is one of the recommended elements for reporting in primary cutaneous melanoma. Per the CAP protocol, regression should be reported as not identified, present (involving <75 or ≥75% of the lesion, if present), or indeterminate.
To date, the biologic significance of melanoma regression has not been completely elucidated. When only a portion of the primary melanoma has undergone regression, it is unclear whether mere reduction in tumor volume has a favorable effect on patient outcome. However, it is conceivable that when only specific subpopulations of the melanoma are eliminated, the degree of aggressiveness of the remaining clones in a patient may predict the patient’s melanoma-specific outcome. In a recent report of 321 cases of thin melanoma (Breslow thickness ≤1.00 mm), Yun et al suggested that one of the factors contributing to poor prognosis associated with regression was the increased dermal lymphatic vessel density and the resulting increased risk of lymphovascular invasion.80
Several studies have failed to show a correlation between regression and patient outcome (Table 1).2, 4, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 In contrast, other studies have indicated that the presence of regression in melanoma was associated with a poor prognosis (Table 2).7, 72, 74, 75, 76, 78, 100, 101, 102, 103 On the other hand, additional studies have suggested that regression indicates a favorable outcome (Table 3).3, 104, 105, 106, 107, 108 Thus, the prognostic significance of regression in primary melanoma has been controversial for many years. In the next section, we discuss some of the most relevant published studies with respect to melanoma recurrence, lymph node metastasis, and survival.
Association between Regression and Recurrence
While comparing only the presence vs absence of late-stage regression, several authors found no correlation between partial regression of melanoma and recurrence. For instance, in one of the largest studies of cutaneous stage I or stage II melanoma (N=7540 cases), Shaw et al noted regression in 67% of 61 stage I melanomas that recurred as well as in 61% of 735 stage I melanomas that did not recur. These data seem to indicate a lack of correlation between regression and recurrence.109 Similar results have been reported by other groups, including two separate studies of 48 melanomas of the extremities (Breslow thickness ≤1.00 mm)82, 84 and a study of 347 cases of melanoma.88 In a recent study of 1349 cutaneous melanomas, however, Morris et al106 showed that regression correlated with decreased local and systemic recurrence.
Association between Regression and Lymph Node Metastases
Some early studies simply comparing the presence or absence of late-stage regression demonstrated that thin melanomas with regression were more prone to metastasis than their counterparts without regression.74, 78, 100, 103 Others have examined the stage and extent of regression and reported increased incidence of metastasis in melanomas with regression.4, 7, 76, 78, 110 In their large study of cutaneous stage I and stage II melanomas, Shaw et al109 reported that regression was present in all 28 cases of thin melanoma (Breslow thickness <0.76 mm) that presented with concurrent regional lymph node metastases (stage II), suggesting an association between regression and lymph node metastasis. In a study by Guitart et al,78 the presence of extensive (>50%) regression on a representative section of melanoma was associated with a higher risk of metastasis. In a study of 103 thin melanomas (Breslow thickness <0.76 mm), Ronan et al76 reported that there was an increased risk for visceral metastasis when more than 77% of the melanoma was regressed. In contrast, some authors showed that the presence of regression in primary melanomas was not associated with lymph node metastasis.2, 86, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97 Some studies have demonstrated a lack of correlation between extent of regression and SLN status.2, 4, 7, 76, 82 Yet other studies have reported that the presence of regression may be associated with negative SLN status.3, 85, 98, 105, 106, 107 Moreover, in a recent meta-analysis of 14 studies that included 10 098 patients with melanoma of various stages, Ribero et al13 demonstrated that regression was associated with lower rates of SLN positivity.
Association between Regression and Survival
In a multifactorial analysis of prognostic variables, Balch et al99 demonstrated that the presence of regression did not affect survival rates in 339 patients with melanoma. In the study of 844 melanomas subdivided into three groups based on Breslow thickness (<0.76, 0.76–1.5, and >1.5 mm), Kelly et al83 reported the overall incidence of late-stage regression to be 20.4% with no difference in 5-year survival rates by lesion thickness. A similar lack of association between regression and survival has been reported by a few other studies.81, 85, 94 In contrast, Slingluff et al72 reported, in a study of 681 thin melanomas (Breslow thickness <0.76 mm), that severe regression in thin melanomas was associated with decreased disease-free interval. Similarly, Sondergaard and Hou-Jensen75 reported a lower 10-year survival rate in patients with past regression and noted that thicker and wider areas of fibrosis were associated with poor survival. In a multivariable logistic regression analysis of 386 cases of clinical stage I melanoma, Clark et al102 reported that the presence of melanoma regression was associated with a lower 8-year survival rate. Furthermore, complete regression was common in thin melanomas with concurrent metastasis and was associated with lower 5-year overall survival rate.4 Only a few studies have reported an association between the presence of regression and better survival rates.104, 108
Notably, most of these studies compared the presence or absence of late-stage regression, while some of the remaining studies compared the extent of regression as focal, partial, or complete or the stage of regression as early vs late or active vs past, with or without an intermediate stage. Only rare studies have examined regression further by measuring the depth of regression-associated fibrosis; Sondergaard and Hou-Jensen75 expanded their analysis of regression by assessing the horizontal width and depth of fibrosis. Similarly, Ronan et al,76 in addition to assessing the stage of regression, examined area and depth of regression. However, no study so far has examined the significance of regression-associated inflammation. These studies highlight the lack of homogeneity among pathologists in the assessment of regression in primary cutaneous melanomas.
In our experience (unpublished data) some histologic features that characterize regression such as the percentage of horizontal extent of regression (based on all sections containing the entire melanoma and not on a representative section) as well as the maximum thickness of regression-associated fibrosis and inflammatory infiltrate may be prognostically informative of the patient’s outcome. Greater depth of regression-associated fibrosis and inflammation and the presence of focal (≤50%) regression significantly correlated with adverse histopathologic prognostic parameters of primary cutaneous melanomas, such as higher Clark level and increased Breslow thickness, mitotic rate, ulceration and lymphovascular invasion. Furthermore, increased thickness of regression-associated fibrosis correlated with reduced overall survival in a multivariate analysis. Similarly, Sondergaard and Hou-Jensen75 have shown that thicker and wider area of regression-associated fibrosis may correlate with poor survival.
Conclusion
Owing to the various inconsistencies in assessing regression, standardization of the evaluation criteria is essential before the biologic and prognostic significance of histologic regression can be recognized.15, 82, 111 Therefore, we recommend that all (dermato)pathologists assess regression in an objective manner and include regression-associated parameters (extent and depth) in their pathology report for primary cutaneous melanomas. We recommend using the following features to diagnose the presence of histologic regression in primary cutaneous melanomas: dermal mononuclear infiltrate composed predominantly of mature lymphocytes, variable degrees of non-laminated dermal fibrosis, melanophagocytosis, telangiectatic blood vessels and variably attenuated epidermis; degeneration of keratinocytes and rarely melanoma cells may also be present. We also recommend assessing the horizontal extent of regression as focal (≤50%) vs intermediate (>50 to ≤75%) vs extensive (>75%) by determining the percentage of horizontal regression in the entire lesion as well as the maximum thickness of regression-associated dermal fibrosis and inflammatory infiltrate with a calibrated ocular micrometer (similar to measuring Breslow thickness). We anticipate that such an objective analysis will provide sufficient clinical evidence over time to understand the prognostic significance of regression and its inclusion in the CAP and AJCC classifications.
References
American Cancer Society Cancer Facts & Figures 2016. American Cancer Society: Atlanta, GA, 2016.
Botella-Estrada R, Traves V, Requena C et al, Correlation of histologic regression in primary melanoma with sentinel node status. JAMA Dermatol 2014; 150: 828–835.
Kaur C, Thomas RJ, Desai N et al, The correlation of regression in primary melanoma with sentinel lymph node status. J Clin Pathol 2008; 61: 297–300.
McClain SE, Shada AL, Barry M et al, Outcome of sentinel lymph node biopsy and prognostic implications of regression in thin malignant melanoma. Melanoma Res 2012; 22: 302–309.
Sumner WC . Spontaneous regression of melanoma. Cancer 1953; 6: 1040–1043.
Ceballos PI, Barnhill RL . Spontaneous regression of cutaneous tumors. Adv Dermatol 1993; 8: 229–261; discussion 262.
Blessing K, McLaren KM . Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology 1992; 20: 315–322.
Bottger D, Dowden RV, Kay PP . Complete spontaneous regression of cutaneous primary malignant melanoma. Plast Reconstr Surg 1992; 89: 548–553.
Emanuel PO, Mannion M, Phelps RG . Complete regression of primary malignant melanoma. Am J Dermatopathol 2008; 30: 178–181.
High WA, Stewart D, Wilbers CR et al, Completely regressed primary cutaneous malignant melanoma with nodal and/or visceral metastases: a report of 5 cases and assessment of the literature and diagnostic criteria. J Am Acad Dermatol 2005; 53: 89–100.
Shai A, Avinoach I, Sagi A . Metastatic malignant melanoma with spontaneous and complete regression of the primary lesion. Case report and review of the literature. J Dermatol Surg Oncol 1994; 20: 342–345.
Smith JL, Stehlin JS . Spontaneous regression of primary malignant melanomas with regional metastases. Cancer 1965; 18: 1399–1415.
Ribero S, Gualano MR, Osella-Abate S et al, Association of histologic regression in primary melanoma with sentinel lymph node status: a systematic review and meta-analysis. JAMA Dermatol 2015; 151: 1301–1307.
Cole WH, Everson TC . Spontaneous regression of cancer: preliminary report. Ann Surg 1956; 144: 366–383.
Requena C, Botella-Estrada R, Traves V et al, Problems in defining melanoma regression and prognostic implication. Actas Dermosifiliogr 2009; 100: 759–766.
Smoller BR . Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol 2006; 19 (Suppl 2): S34–S40.
Zurac S, Negroiu G, Petrescu S et al, Spectrum of morphologic alterations of regression in cutaneous melanoma—potential for improving disease prognosis. Rom J Intern Med 2012; 50: 145–153.
Nathanson . Spontaneous regression of malignant melanoma: a review of the literature on incidence, clinical features, and possible mechanisms. Natl Cancer Inst Monogr 1976; 44: 67–76.
Bayer-Garner IB, Ivan D, Schwartz MR et al, The immunopathology of regression in benign lichenoid keratosis, keratoacanthoma and halo nevus. Clin Med Res 2004; 2: 89–97.
Cook MG . The significance of inflammation and regression in melanoma. Virchows Arch A Pathol Anat Histopathol 1992; 420: 113–115.
Saleh FH, Crotty KA, Hersey P et al, Primary melanoma tumour regression associated with an immune response to the tumour-associated antigen melan-A/MART-1. Int J Cancer 2001; 94: 551–557.
Tarhini AA . Immunotherapy of melanoma. Curr Mol Pharmacol 2016; 9: 196–207.
Hughes T, Klairmont M, Sharfman WH et al, Interleukin-2, ipilimumab, and anti-PD-1: clinical management and the evolving role of immunotherapy for the treatment of patients with metastatic melanoma. Cancer Biol Ther 2015.
Elston DM . Mechanisms of regression. Clin Med Res 2004; 2: 85–88.
Bernsen MR, Diepstra JH, van Mil P et al, Presence and localization of T-cell subsets in relation to melanocyte differentiation antigen expression and tumour regression as assessed by immunohistochemistry and molecular analysis of microdissected T cells. J Pathol 2004; 202: 70–79.
Janeway CA, Travers P, Walport M et al(eds). The major histocompatibility complex and its functionsImmunobiology: The Immune System in Health and Disease, 5th edn. Garland Science: New York, 2001.
Janeway CA, Travers P, Walport M et al, (eds). Using the immune response to attack tumorsImmunobiology: The Immune System in Health and Disease, 5th edn. Garland Science: New York, 2001.
Smith C, Cerundolo V . Immunotherapy of melanoma. Immunology 2001; 104: 1–7.
Stromnes IM, Schmitt TM, Chapuis AG et al, Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol Rev 2014; 257: 145–164.
Hofmann O, Caballero OL, Stevenson BJ et al, Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci USA 2008; 105: 20422–20427.
Traversari C, van der Bruggen P, Luescher IF et al, A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med 1992; 176: 1453–1457.
Escors D . Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J Sci 2014; 2014: 1–25.
Nicholaou T, Ebert L, Davis ID et al, Directions in the immune targeting of cancer: lessons learned from the cancer-testis Ag NY-ESO-1. Immunol Cell Biol 2006; 84: 303–317.
Robbins PF, Morgan RA, Feldman SA et al, Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29: 917–924.
Vigneron N . Human tumor antigens and cancer immunotherapy. Biomed Res Int 2015; 2015: 948501.
Haanen JB . Immunotherapy of melanoma. EJC Suppl 2013; 11: 97–105.
Printz C . Spontaneous regression of melanoma may offer insight into cancer immunology. J Natl Cancer Inst 2001; 93: 1047–1048.
McGovern VJ . Spontaneous regression of melanoma. Pathology 1975; 7: 91–99.
Kostner AH, Johansen RF, Schmidt H et al, Regression in cancer following fever and acute infection. Acta Oncol 2013; 52: 455–457.
Tran T, Burt D, Eapen L et al, Spontaneous regression of metastatic melanoma after inoculation with tetanus-diphtheria-pertussis vaccine. Curr Oncol 2013; 20: e270–e273.
Dunn GP, Bruce AT, Ikeda H et al, Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002; 3: 991–998.
Ichim CV . Revisiting immunosurveillance and immunostimulation: implications for cancer immunotherapy. J Transl Med 2005; 3: 8.
Kim KD, Zhao J, Auh S et al, Adaptive immune cells temper initial innate responses. Nat Med 2007; 13: 1248–1252.
Lister JA, Capper A, Zeng Z et al, A conditional zebrafish MITF mutation reveals MITF levels are critical for melanoma promotion vs regression in vivo. J Invest Dermatol 2014; 134: 133–140.
Bastian BC . Hypothesis: a role for telomere crisis in spontaneous regression of melanoma. Arch Dermatol 2003; 139: 667–668.
Dranoff G . Experimental mouse tumour models: what can be learnt about human cancer immunology? Nat Rev Immunol 2011; 12: 61–66.
Pardoll DM . The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264.
Kuzu OF, Nguyen FD, Noory MA et al, Current state of animal (mouse) modeling in melanoma research. Cancer Growth Metastasis 2015; 8 (Suppl 1): 81–94.
Bosenberg M, Muthusamy V, Curley DP et al, Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 2006; 44: 262–267.
Beaumont KA, Mohana-Kumaran N, Haass NK . Modeling Melanoma In Vitro and In Vivo. Healthcare (Basel) 2013; 2: 27–46.
Murphy GF, Wilson BJ, Girouard SD et al, Stem cells and targeted approaches to melanoma cure. Mol Aspects Med 2014; 39: 33–49.
Schatton T, Frank MH . Antitumor immunity and cancer stem cells. Ann NY Acad Sci 2009; 1176: 154–169.
Frank NY, Schatton T, Frank MH . The therapeutic promise of the cancer stem cell concept. J Clin Invest 2010; 120: 41–50.
Schatton T, Schutte U, Frank NY et al, Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res 2010; 70: 697–708.
Sandru A, Voinea S, Panaitescu E et al, Survival rates of patients with metastatic malignant melanoma. J Med Life 2014; 7: 572–576.
Bhatia S, Tykodi SS, Thompson JA . Treatment of metastatic melanoma: an overview. Oncology (Williston Park) 2009; 23: 488–496.
Overwijk WW, Theoret MR, Restifo NP . The future of interleukin-2: enhancing therapeutic anticancer vaccines. Cancer J Sci Am 2000; 6 (Suppl 1): S76–S80.
Tarhini AA, Gogas H, Kirkwood JM . IFN-alpha in the treatment of melanoma. J Immunol 2012; 189: 3789–3793.
Kirkwood JM, Richards T, Zarour HM et al, Immunomodulatory effects of high-dose and low-dose interferon alpha2b in patients with high-risk resected melanoma: the E2690 laboratory corollary of intergroup adjuvant trial E1690. Cancer 2002; 95: 1101–1112.
Parlato S, Santini SM, Lapenta C et al, Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood 2001; 98: 3022–3029.
Fuertes MB, Kacha AK, Kline J et al, Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011; 208: 2005–2016.
Davar D, Kirkwood JM . Adjuvant therapy of melanoma. Cancer Treat Res 2016; 167: 181–208.
Redman JM, Gibney GT, Atkins MB . Advances in immunotherapy for melanoma. BMC Med 2016; 14: 20.
Francisco LM, Sage PT, Sharpe AH . The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010; 236: 219–242.
Rabinovich GA, Gabrilovich D, Sotomayor EM . Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 2007; 25: 267–296.
Chambers CA, Kuhns MS, Egen JG et al, CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001; 19: 565–594.
Johnson DB, Lovly CM, Sullivan RJ et al, Melanoma driver mutations and immune therapy. Oncoimmunology 2016; 5: e1051299.
He J, Hu Y, Hu M et al, Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep 2015; 5: 13110.
Wang X, Teng F, Kong L et al, PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther 2016; 9: 5023–5039.
Novotny JF, Cogswell J, Inzunza H et al, Establishing a complementary diagnostic for anti-PD-1 immune checkpoint inhibitor therapy. Ann Oncol 2016; 27: 1966–1969.
Kang S, Barnhill RL, Mihm MC et al, Histologic regression in malignant melanoma: an interobserver concordance study. J Cutan Pathol 1993; 20: 126–129.
Slingluff CL Jr., Vollmer RT, Reintgen DS et al, Lethal ‘thin’ malignant melanoma. Identifying patients at risk. Ann Surg 1988; 208: 150–161.
Massi G, LeBoit P . Regressing and regressed melanoma, In: Massi G, LeBoit P (eds). Histological Diagnosis of Nevi and Melanoma, 2nd edn. Springer: Heidelberg, 2014, pp 699–720.
Naruns PL, Nizze JA, Cochran AJ et al, Recurrence potential of thin primary melanomas. Cancer 1986; 57: 545–548.
Sondergaard K, Hou-Jensen K . Partial regression in thin primary cutaneous malignant melanomas clinical stage I. A study of 486 cases. Virchows Arch A Pathol Anat Histopathol 1985; 408: 241–247.
Ronan SG, Eng AM, Briele HA et al, Thin malignant melanomas with regression and metastases. Arch Dermatol 1987; 123: 1326–1330.
Byers HR, Bhawan J . Pathologic parameters in the diagnosis and prognosis of primary cutaneous melanoma. Hematol Oncol Clin North Am 1998; 12: 717–735.
Guitart J, Lowe L, Piepkorn M et al, Histological characteristics of metastasizing thin melanomas: a case-control study of 43 cases. Arch Dermatol 2002; 138: 603–608.
Crowson AN, Magro CM, Mihm MC . Prognosticators of melanoma, the melanoma report, and the sentinel lymph node. Mod Pathol 2006; 19 (Suppl 2): S71–S87.
Yun SJ, Gimotty PA, Hwang WT et al, High lymphatic vessel density and lymphatic invasion underlie the adverse prognostic effect of radial growth phase regression in melanoma. Am J Surg Pathol 2011; 35: 235–242.
McGovern VJ, Shaw HM, Milton GW . Prognosis in patients with thin malignant melanoma: influence of regression. Histopathology 1983; 7: 673–680.
Cooper PH, Wanebo HJ, Hagar RW . Regression in thin malignant melanoma. Microscopic diagnosis and prognostic importance. Arch Dermatol 1985; 121: 1127–1131.
Kelly JW, Sagebiel RW, Blois MS . Regression in malignant melanoma. A histologic feature without independent prognostic significance. Cancer 1985; 56: 2287–2291.
Wanebo HJ, Cooper PH, Hagar RW . Thin (less than or equal to 1 mm) melanomas of the extremities are biologically favorable lesions not influenced by regression. Ann Surg 1985; 201: 499–504.
Brogelli L, Reali UM, Moretti S et al, The prognostic significance of histologic regression in cutaneous melanoma. Melanoma Res 1992; 2: 87–91.
Wagner JD, Gordon MS, Chuang TY et al, Predicting sentinel and residual lymph node basin disease after sentinel lymph node biopsy for melanoma. Cancer 2000; 89: 453–462.
Fontaine D, Parkhill W, Greer W et al, Partial regression of primary cutaneous melanoma: is there an association with sub-clinical sentinel lymph node metastasis? Am J Dermatopathol 2003; 25: 371–376.
Topping A, Dewar D, Rose V et al, Five years of sentinel node biopsy for melanoma: the St George’s Melanoma Unit experience. Br J Plast Surg 2004; 57: 97–104.
Kruper LL, Spitz FR, Czerniecki BJ et al, Predicting sentinel node status in AJCC stage I/II primary cutaneous melanoma. Cancer 2006; 107: 2436–2445.
Cecchi R, Pavesi M, Buralli L et al, Tumour regression does not increase the risk of sentinel node involvement in thin melanomas. Chir Ital 2008; 60: 257–260.
Gutzmer R, Satzger I, Thoms KM et al, Sentinel lymph node status is the most important prognostic factor for thick (>or=4 mm) melanomas. J Dtsch Dermatol Ges 2008; 6: 198–203.
Mandala M, Imberti GL, Piazzalunga D et al, Clinical and histopathological risk factors to predict sentinel lymph node positivity, disease-free and overall survival in clinical stages I-II AJCC skin melanoma: outcome analysis from a single-institution prospectively collected database. Eur J Cancer 2009; 45: 2537–2545.
Socrier Y, Lauwers-Cances V, Lamant L et al, Histological regression in primary melanoma: not a predictor of sentinel lymph node metastasis in a cohort of 397 patients. Br J Dermatol 2010; 162: 830–834.
Burton AL, Gilbert J, Farmer RW et al, Regression does not predict nodal metastasis or survival in patients with cutaneous melanoma. Am Surg 2011; 77: 1009–1013.
Callender GG, Egger ME, Burton AL et al, Prognostic implications of anatomic location of primary cutaneous melanoma of 1 mm or thicker. Am J Surg 2011; 202: 659–664; discussion 664-655.
Tejera-Vaquerizo A, Nagore E, Herrera-Acosta E et al, Prediction of sentinel lymph node positivity by growth rate of cutaneous melanoma. Arch Dermatol 2012; 148: 577–584.
Grotz TE, Vaince F, Hieken TJ . Tumor-infiltrating lymphocyte response in cutaneous melanoma in the elderly predicts clinical outcomes. Melanoma Res 2013; 23: 132–137.
Han D, Zager JS, Shyr Y et al, Clinicopathologic predictors of sentinel lymph node metastasis in thin melanoma. J Clin Oncol 2013; 31: 4387–4393.
Balch CM, Murad TM, Soong SJ et al, A multifactorial analysis of melanoma: prognostic histopathological features comparing Clark’s and Breslow’s staging methods. Ann Surg 1978; 188: 732–742.
Gromet MA, Epstein WL, Blois MS . The regressing thin malignant melanoma: a distinctive lesion with metastatic potential. Cancer 1978; 42: 2282–2292.
Paladugu RR, Yonemoto RH . Biologic behavior of thin malignant melanomas with regressive changes. Arch Surg 1983; 118: 41–44.
Clark WH Jr, Elder DE, Guerry Dt et al, Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst 1989; 81: 1893–1904.
Olah J, Gyulai R, Korom I et al, Tumour regression predicts higher risk of sentinel node involvement in thin cutaneous melanomas. Br J Dermatol 2003; 149: 662–663.
Trau H, Kopf AW, Rigel DS et al, Regression in malignant melanoma. J Am Acad Dermatol 1983; 8: 363–368.
Liszkay G, Orosz Z, Peley G et al, Relationship between sentinel lymph node status and regression of primary malignant melanoma. Melanoma Res 2005; 15: 509–513.
Morris KT, Busam KJ, Bero S et al, Primary cutaneous melanoma with regression does not require a lower threshold for sentinel lymph node biopsy. Ann Surg Oncol 2008; 15: 316–322.
Testori A, De Salvo GL, Montesco MC et al, Clinical considerations on sentinel node biopsy in melanoma from an Italian multicentric study on 1313 patients (SOLISM-IMI). Ann Surg Oncol 2009; 16: 2018–2027.
Ribero S, Osella-Abate S, Sanlorenzo M et al, Favourable prognostic role of regression of primary melanoma in AJCC stage I-II patients. Br J Dermatol 2013; 169: 1240–1245.
Shaw HM, McCarthy SW, McCarthy WH et al, Thin regressing malignant melanoma: significance of concurrent regional lymph node metastases. Histopathology 1989; 15: 257–265.
Bertolli E, de Macedo MP, Pinto CA et al, Evaluation of melanoma features and their relationship with nodal disease: the importance of the pathological report. Tumori 2015; 101: 501–505.
Sagebiel R . Regression and other factors of prognostic interest in malignant melanoma. Arch Dermatol 1985; 121: 1125–1126.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This review examines the biologic mechanisms underlying regression in primary cutaneous melanomas, with emphasis on the role of immunotherapy. It also summarizes the conflicting data on the potential significance of histologic assessment of regression in predicting clinical outcome of melanoma patients, such as recurrence and metastasis.
Rights and permissions
About this article

Cite this article
Aung, P., Nagarajan, P. & Prieto, V. Regression in primary cutaneous melanoma: etiopathogenesis and clinical significance. Lab Invest 97, 657–668 (2017). https://doi.org/10.1038/labinvest.2017.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.8
This article is cited by
-
Strategies for improving the performance of prediction models for response to immune checkpoint blockade therapy in cancer
BMC Research Notes (2024)
-
Risk factors and prognosis of orbital exenteration in conjunctival melanoma
Eye (2023)
-
Deciphering the immune reaction leading to spontaneous melanoma regression: initial role of MHCII+ CD163− macrophages
Cancer Immunology, Immunotherapy (2023)
-
Mind your head: two cases of mucosal metastasis of BRAF-mutated melanoma of the scalp
Virchows Archiv (2022)
-
Histological regression in melanoma: impact on sentinel lymph node status and survival
Modern Pathology (2021)
