
Abstract
We previously reported that long-term treatment with a calcineurin inhibitor impairs autophagy process in pancreatic beta cells. This study investigated the effect of Korean red ginseng extract (KRGE) on autophagy modulated by oxidative stress. In mice with tacrolimus (Tac)-induced diabetes mellitus, KRGE alleviated islet dysfunction and decreased oxidative stress and autophagic vacuoles. In vitro, KRGE decreased autophagosome formation and attenuated lysosomal degradation, accompanied by improved beta cell viability and insulin secretion. Addition of 3-methyladenine (3-MA), an inhibitor of autophagosomes, to KRGE further improved cell viability and insulin secretion, and bafilomycin A (BA), an inhibitor of lysosomal function, reduced the effects of KRGE. At the subcellular level, Tac caused mitochondrial dysfunction (impaired mitochondrial oxygen consumption, ATP production, and increased reactive oxygen species production). But KRGE improved these parameters. The effect of KRGE on mitochondrial function enhanced by 3-MA but decreased by BA, suggesting a causal relationship between KRGE effect and autophagy modulation in Tac-induced mitochondrial dysfunction. These findings indicate that KRGE modulates autophagy favorably by reducing Tac-induced oxidative stress, and this effect is closely associated with improvement of mitochondrial function.
Similar content being viewed by others


Main
Tacrolimus (Tac) is widely used as a maintenance immunosuppressant in renal transplant recipients (KRTs) but it causes considerable metabolic abnormalities. Particularly, new-onset diabetes after transplantation (NODAT) occurs in 10–25% of the patients receiving Tac,1, 2 and NODAT reduces graft survival and increase the risk of infectious and cardiovascular diseases.3 The pathogenesis of Tac-induced diabetes mellitus (DM) is unclear, but we and others reported that the direct toxic effect of Tac on pancreatic beta cells and oxidative stress has a pivotal role in Tac-induced DM.4, 5, 6
Ginseng, a traditional medicine, is commonly used to modulate immune functions and metabolism, and ginsenosides are the pharmacologically active components in ginseng and that are used as traditional medicine to treat various diseases.7 Korean red ginseng extract (KRGE) is a popular and commercially available health food, but its mechanism is still undetermined.
Therefore, our initial study focused on effectiveness and safety in kidney transplant recipients, and we reported that KRGE is immunologically and pharmacologically safe and provides the benefit of reducing oxidative stress in cyclosporine-induced nephrotoxicity or pancreatic islet injury.8, 9, 10 Recently, we found that KRGE effect is closely associated with autophagy modulation in chronic cyclosporine nephrotoxicity.11 In this study, we investigated whether ginseng effect on autophagy modulation can be observed in Tac-induced oxidative stress in pancreatic islet cells, and whether mitochondria are involved in this process.
Materials and methods
Animals and Drugs
Eight-week-old male Balb/c mice (Charles River Technology, Seoul, Korea) were housed in a controlled-light setting at the animal facility of the Catholic University of Korea. The Animal Care and Use Committee of the Catholic University of Korea approved the experimental protocol (CUMC-2016-0223-01). All procedures performed in this study were in accordance with the ethical guidelines for animal studies. Mice were maintained on a 12/12 h light/dark cycle, fed a 0.01% salt diet (AIN-93G; Research Diets), and provided ad libitum access to water. Tac (Prograft, Astellas Pharma, Ibaraki, Japan) was diluted in olive oil (Sigma-Aldrich, St. Louis, MO, USA) to a final concentration of 1.5 mg/kg. KRGE (Korea Ginseng Corporation, Seoul, Korea) was diluted in sterile water. According to the manufacturer’s data, the main components of the KRGE were Rg1 (2.01%), Rb1 (8.27%), Rg3 (s) (1.04%), Re (2.58%), Rc (3.90%), Rb2 (3.22%), Rd (1.09%), Rf (1.61%), Rh1 (0.95%), and Rg2 (s) (1.35%).
Experimental Study for In Vivo
After acclimatization for 1 week, weight-matched mice were randomized to four groups (n=8 each) and treated daily with a subcutaneous injection of 1.5 mg/kg Tac or 1 ml/kg Vh (olive oil; O1514; Sigma-Aldrich) with or without KRGE (0.4 g/kg, oral gavage; Korea Ginseng) for 4 weeks. Routes of administration and doses of drugs were chosen based on previous studies.4, 8, 11 Mice were pair-fed and their body weights were monitored daily. After the 4-week treatment, mice were housed individually in metabolic cages (Tecniplast, Buguggiate, Italy) for the measurement of urine volume over 24 h. The following day, mice were anesthetized for blood collection and killed for tissue specimen collection. Serum creatinine levels and trough levels of Tac in the blood were measured using commercial enzyme-linked immunosorbent assay (ELISA) kits (Nova Lifetech, Mongkok Ki, Hong Kong).
Pancreatic Function and Islet Size
An intraperitoneal glucose tolerance test (IPGTT) was performed at the end of the 4-week treatment period. Briefly, after 1 day of fasting, 50% dextrose (1.5 g/kg) was administered to mice. The blood glucose concentration was measured just before and at 30, 60, 90, and 120 min after dextrose administration using a glucose analyzer (Accu-Check; Roche Diagnostics, Basel, Switzerland). The area under the curve of glucose (AUCg) was calculated by trapezoidal estimation from the concentrations obtained in the IPGTT. The fasting serum insulin level was measured using a sandwich ELISA (Millipore Corporation, St. Charles, MO, USA). For determination of islet size, the numbers of beta cells were quantified in captured immunohistochemistry images for insulin detection (TDI Scope Eye, Version 3.6 for Windows; Seoul, Korea). Insulin-positive cells were quantified by counting cells in ~20 randomly selected non-overlapping islets for all eight mice in each group.
Antibodies
Primary antibodies used for confocal microscopy or immunoblot analysis were as follows: anti-insulin (I2018, Sigma-Aldrich; 18-0067, Invitrogen); anti-8-OHdG (MOG-020P; Japan Institute for the Control of Aging); anti-LC3B (L7543, Sigma-Aldrich; ALX-803-080, ENZO Life Sciences); anti-LAMP-2A (ab18528, Abcam); and anti-DDK (TA50011-100, OriGene Technologies).
Detection of 8-OHdG
Oxidative DNA damage was evaluated based on the level of 8-OHdG in serum and tissue sections using either competitive ELISA (Cell Biolabs, San Diego, CA, USA) or anti-8-OHdG antibody for immunostaining.
Cell Culture for In Vitro
The rat insulinoma cell line INS-1 was grown in RPMI-1640 medium supplemented with 10 nM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 50 mM 2-mercaptoethanol (all from Sigma-Aldrich), 100 IU/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum (all from Wisent Bio, Saint-Bruno, QC, Canada) in a humidified 5% CO2 atmosphere. Cells were seeded in culture plates and treated with Tac (40 μg/ml) or KRGE (100 pg/ml) with or without 3-methyladenine (3-MA; 10 mM; M9281; Sigma-Aldrich) or bafilomycin A (BA; 2 nM; B1793; Sigma-Aldrich) for 6 h.
Cell Viability
Cell viability measured using cell-counting kit-8 (CCK-8, CK04; Dojindo Molecular Technologies, Rockville, MD, USA) assay kit. One day after seeding, INS-1 cells were subjected to various treatments for the specified periods. Before the end of the treatments, CCK-8 or propidium iodide (PI; 556463; BD Biosciences, San Jose, CA, USA) solution was added to each well or trypsinized cells according to the manufacture’s protocol. After addition of CCK-8, absorbance was measured at 450 nm using a Versa Max ELISA Reader (Molecular Devices, Sunnyvale, CA, USA). The PI-stained dead cells were detected using a FACS Calibur flow cytometer (BD Biosciences).
Glucose-Stimulated Insulin Release
To measure glucose-stimulated insulin release (GSIS), cells were seeded in 24-well plates at a density of 1 × 106/well for 48 h and subjected to various treatments for the specified periods. After treatment, supernatant was collected as a basal medium and the cells were washed with Krebs–Ringer modified buffer (KRB; 2 g/l NaHCO3, 6.7 g/l NaCl, 0.373 g/l KCl, 0.368 g/l CaCl2, 0.2 g/l MgCl2, and 5.95 g/l HEPES). Subsequently, the cells were incubated with KRB containing 25 mM glucose for 60 min. The supernatant was sampled to measure the insulin concentration using commercial ELISA kit (Millipore Corporation). Insulin values were then normalized to the protein content of each sample.
Cell Transfection
INS-1 cells were transfected with DDK-LC3B plasmids (RR209172, OriGene Technologies) using Lipofectamine 2000 (Invitrogen) according to the manufacturers’ instructions. The cells were incubated with Tac (40 μg/ml) with or without KRGE (100 pg/ml) for 6 h. To determine autophagosome–lysosome fusion, double immunofluorescence was then performed for detecting DDK and LAMP-2A (lysosome marker). Subsequently, the localization of LC3B and LAMP-2A was observed using a Zeiss LSM700 confocal microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany).
Immunoblot Analysis
Cells were lysed in PRO-PREP protein extraction solution (17081; Intron Biotechnology) according to the manufacturer’s instructions. Equal amounts of protein were subjected to immunoblotting analysis with primary antibodies. Signals were detected using an enhanced chemiluminescence system (ATTO, Tokyo, Japan). Quantification was performed using relative densities, with the control group set at 100%. Band densities were normalized to that of the beta-actin bands from the same gel (Quantity One version 4.4.0; Bio-Rad, Hercules, CA, USA).
Transmission Electron Microscopy
After fixation in 2.5% glutaraldehyde (in 0.1 M phosphate buffer), pancreatic tissues were postfixed with 1% OsO4 and embedded in Epon 812. Ultrathin sections were cut, stained with uranyl acetate/lead citrate, and photographed with a JEM-1200EX transmission electron microscope (JEOL, Tokyo, Japan). Sections were scanned randomly at 20 different sites per sample at × 5000 magnification. The numbers and areas of autophagic vacuoles (autophagosomes and autolysosomes) per cell in the scanned areas were measured using an imaging software (TDI Scope Eye).
LysoTracker Staining
LysoTracker Red uses the accumulation of a cationic fluorescent dye in acidic cellular compartments as an indicator of total lysosome content. One day after seeding, the cells were treated with each drug in serum-free RPMI medium. After 6 h, the drug-containing medium was removed and the cells were incubated with 100 nM LysoTracker Red DND-99 (L7529; Molecular Probes) for 1 h at 37 °C. Subsequently, the cells were analyzed using FACS Calibur flow cytometer or confocal microscope.
Mitochondrial ROS Detection
After treatment with drugs or vehicle, cells were incubated with MitoSOX red (M36008; Invitrogen) for 30 min at 37 °C according to the manufacturer’s instruction and then analyzed using FACS Calibur flow cytometer or Zeiss LSM700 confocal microscope for detection of mitochondrial ROS (superoxide anion). Data were collected in forward scatter and side scatter, and a total of 10 000 events were collected for each sample.
Determination of Mitochondrial Damage
Mitochondrial integrity was assessed with MitoTracker Deep Red FM (M22426; Invitrogen) fluorescent probe. These probes do not fluoresce until they enter live cells, where they are oxidized to the fluorescent mitochondrion-selective probe and then sequestered in the mitochondria. Cells were first treated with drugs or vehicle for 20 min at 37 °C according to the manufacturer’s instruction and then analyzed using a FACS Calibur flow cytometer or Zeiss LSM700 confocal microscope.
Mitochondrial Membrane Potential (ΔΨm)
ΔΨm of INS-1 cells was measured using JC-1, a fluorescent, lipophilic, and cationic probe (10009172; Cayman Chemical). JC-1 permeates the plasma membrane of live cells; its uptake into mitochondria is driven by the ΔΨm. Functional mitochondria are polarized. JC-1 is rapidly taken up in polarized mitochondria. This uptake raises the concentration of JC-1 in mitochondria leading to the formation of aggregates (J-aggregates). Excitation of J-aggregates by a 488 nm laser provokes a red spectral shift emission (590 nm). JC-1 does not accumulate in depolarized mitochondria and remains in the cytoplasm as monomers, which emit green fluorescence (525 nm). Thus, in healthy cells, JC-1 fluorescence can be observed in both the green (FL-1) and red (FL-2) channels. A loss of red fluorescence indicates depolarized mitochondria. After treatment with drugs or vehicle, cells were incubated with JC-1 staining solution for 20 min at 37°C according to the manufacturer’s instruction and then analyzed using a FACS Calibur flow cytometer or Zeiss LSM700 confocal microscope.
OCR Experiments
The rate of oxygen consumption of cells was assessed real-time with the XF24 Extracellular Flux Analyzer (Seahorse Biosciences, Billerica, MA, USA). After treatment with drugs or vehicle, the cells were transferred to running media (102353-100; Seahorse Biosciences; DMEM-based medium supplemented with 5.5 mM glucose, 1 mM sodium pyruvate, and 4 mM l-glutamine, pH 7.4, 37 °C) and incubated at 37°C in a non-CO2 incubator for 1 h. Mitochondrial inhibitors were used as modulators to determine several bioenergetic and mitochondrial function parameters, including basal respiration, non-mitochondrial respiration, proton leakage, maximal respiration, spare respiratory capacity, and ATP production according to methods reported by Dott et al.12 The mitochondrial inhibitors used were the ATP synthase inhibitor oligomycin (1 μM), FCCP (2 μM), and the complex I and III inhibitor rotenone/antimycin A (0.5 μM). OCR values were then normalized to the protein content of each sample. Three to four wells were used for each experimental group.
Statistical Analysis
Data are expressed as the mean±s.e. of at least three independent experiments. Multiple comparisons between groups were performed by one-way analysis of variance with Bonferroni post hoc tests using IBM SPSS Statistics (Version 24; IBM, Armonk, NY, USA). Results with P-values of <0.05 were considered significant.
Results
KRGE Improves Pancreatic Islet Function and Autophagy in an Experimental Model of Tac-Induced DM
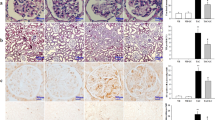
Mice treated with Tac showed overt DM, demonstrated by increased AUCg and lower levels of plasma insulin as compared with the Vh group. Concomitant treatment with KRGE reversed these changes (Figures 1a–c; Table 1). KRGE also increased the islets size reduced by Tac (Figure 1d). Tac increased 8-OHdG levels in the pancreatic islet and serum compared with the Vh group, but KRGE reduced these levels compared with the Tac group (Figures 1e and f). TEM showed markedly reduced numbers of insulin granules in Tac group compared with the Vh group but KRGE recovered the number of insulin granules. (Figure 2). High-magnification TEM (Figure 2c) showed that increased number and areas of autophagic vacuoles/vesicles in the Tac group was decreased with KRGE treatment.

Administration of KRGE reduced Tac-induced diabetes mellitus in mice. Blood glucose levels measured using IPGTTs (a), calculation of the AUCg from IPGTTs (b), and plasma insulin levels (c). (d) Quantitative estimation of insulin-positive area per islet. Oxidative stress was measured by 8-OHdG level in the islets (e) and serum (f). The data are presented as means±s.e.’s. N=8 per group. #P<0.05 vs Vh group; $P<0.05 vs Vh+KRGE group; @P<0.05 vs Tac group.

Transmission electron microscopy of pancreatic beta cells of mice with Tac-induced diabetes mellitus. (a, b) Representative images and quantitative estimation of insulin granules (arrowheads) in the cytoplasm. Scale bar=1 μm. (c) Higher-magnification images from the Tac-treated group, showing organelles surrounded by double membranes representing autophagosomes (double arrows). Scale bar=100 or 200 nm. (d, e) Numbers and areas of autophagic vacuoles containing autophagosome and autolysosomes. Asterisk indicates mitochondria in autophagosome. The data are presented as means±s.e.’s. N=8 per group. #P<0.05 vs Vh group; @P<0.05 vs Tac group.
KRGE has a Direct Protective Effect Against Tac-Induced Pancreatic Islet Injury
The aim of this study was to investigate whether KRGE has direct protective effect against Tac-induced injury in INS-1 cells. Tac decreased INS-1 cell viability compared to that of the untreated control cells, but KRGE significantly improved cell viability (Figure 3a). PI staining followed by flow cytometry showed that Tac increased but KRGE significantly decreased the cell death incidence (Figures 3b and c). In addition to cell viability, KRGE improved beta cell function measured with GSIS. KRGE significantly increased insulin secretion compared with Tac groups, suggesting preserving effect of KRGR on pancreatic islet function (Figure 3d).

Effect of KRGE treatment on Tac-induced INS-1 cell injury. (a) Cell viability using CCK-8. (b, c) Representative flow cytometric dot plots showing the side-scattered light (SSC) (cell granularity)/propidium Iodide (PI) gate and quantitative estimation of the percentage of PI-positive cells, respectively. (d) The insulin level in media secreted in response to glucose determined by glucose-stimulated insulin secretion (GSIS) test. The data are presented as means±s.e.’s of at least three independent experiments. #P<0.05 vs the control group; $P<0.05 vs KRGE group; @P<0.05 vs Tac group.
KRGE Reduces Autophagosome Burden and Activates Autophagosome Clearance in Tac-Induced Pancreatic Islet Injury
To determine whether KRGE modulates Tac-induced autophagy, we evaluated the influence of KRGE on Tac-induced autophagosome formation and clearance in INS-1 cells. Tac significantly increased but KRGE decreased LC3B-II/LC3B-I ratio, suggesting attenuation of autophagosome accumulation by KRGE (Figures 4a and b). Next, we measured the activation of lysosomal function with LysoTracker red and flow cytometry (Figures 4c and d). Tac significantly increased lysosomal pH, whereas KRGE reversed this change. Next, we evaluated the autophagosome–lysosome fusion by double immunofluoresnce. Compared to treatment with Tac, KRGE significantly reduced punctate LC3B expression (Figure 4f) and enhanced colocalization of LC3B and LAMP-2A (yellow fluorescence; Figures 4e and g).

Effect of KRGE on Tac-induced impairment of autophagy in INS-1 cells. (a, b) Representative immunoblot and quantification of LC3B-II in Tac- and KRGE-treated INS-1 cells. The optical densities of bands in each lane were normalized to beta-actin optical density from the same gel. (c, d) Lysosomal acidification evaluated in the cells treated with Tac and KRGE using LysoTracker red. Fluorescence intensity was detected by flow cytometry. (e) Representative confocal immunofluorescence images of DDK-LC3B (autophagosome marker, red) and LAMP-2A (lysosome marker, green). DDK-LC3B was transfected into the cells. The cells were then treated with Tac and/or KRGE. LC3B punctate staining was detected by immunostaining with anti-DDK antibody. Arrowheads indicate double-positive fluorescence (yellow). Nuclear counterstaining was performed with DAPI (blue). (f) Quantitative analysis of LC3B functa per cell. (g) Numbers of LC3B-positive lysosomes. Scale bar=10 μM. The data are presented as means±s.e.’s of at least three independent experiments. #P<0.05 vs the control group; $P<0.05 vs KRGE group; @P<0.05 vs Tac group.
To evaluate protective effect of KREG causally associated with autophagy regulation, we added inhibitor of autophagosome formation or lysosomal inhibitor. As shown in Figure 5, 3-MA improved cell viability and insulin secretion, and reduced cell death under Tac treatment, and this effect was further enhanced by KRGE. Addition of BA abolished increased cell viability and insulin secretion by KRGE (Figures 5a and b).

Influence of autophagy on INS-1 cell injury during Tac and KRGE treatment. INS-1 cells were treated with either 3-methyladenine (3-MA, an inhibitor of autophagosome formation) or bafilomycin A1 (BA, a specific inhibitor of the vacuolar type H+-ATPase in lysosomes). The effect of 3-MA or BA on cell viability, percentage of PI-positive cells, and glucose-stimulated insulin secretion (GSIS) was evaluated using CCK-8 assay (a), flow cytometry (b), and GSIS test (c), respectively. The data are presented as means±s.e.’s of at least three independent experiments. #P<0.05 vs the control group; $P<0.05 vs Tac group; *P<0.05 vs Tac+3-MA group; @P<0.05 vs Tac+KRGE group; &P<0.05 vs Tac+KRGE+3-MA group.
Effect of KRGE on Tac-Induced Mitochondrial Dysfunction and ROS Production
The number of healthy mitochondria in living cells was evaluated with cell-permeant MitoTracker Red probes followed by flow cytometric detection. The reduced number of MitoTracker-stained cells in Tac group was significantly increased by KRGE (Figures 6a and b). In addition, KRGE significantly reduced the Tac-induced MitoSOX Red fluorescence and have protective effect via autophagy after treatment with either 3-MA or BA (Figures 6c and d). KRGE increased polarized cells population, whereas decreased higher depolarized cells population compared with Tac alone as shown in Figure 7. The rate of oxygen consumption of cells was assessed as shown in Figure 8. The KRGE significantly increased basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity compared with the Tac group (Figures 8b–e). To investigate the association of OCR with autophagy, the OCR of either 3-MA- or BA-treated cells was compared with that of Tac+KRGE group.

Effect of KRGE on Tac-induced mitochondrial damage and ROS production in INS-1 cells. (a, b) MitoTracker staining for detecting mitochondria using flow cytometric analysis. (c, d) Representative flow cytometric dot plots showing the side scatter (SSC) (cell granularity)/MitoSOX Red gate and quantitative estimation of the percentage of MitoSOX Red-positive cells. BA, bafilomycin A1; 3-MA, 3-methyladenine. The data are presented as means±s.e.’s of at least three independent experiments. #P<0.05 vs the control group; $P<0.05 vs Tac group; *P<0.05 vs Tac+3-MA group; @P<0.05 vs Tac+KRGE group; &P<0.05 vs Tac+KRGE+3-MA group.

KRGE reduces Tac-induced impairment of mitochondrial membrane potential in INS-1 cells. (a) Mitochondrial membrane potential (ΔΨm) was determined by flow cytometric analysis with JC-1- labeling. (b, c) Representative flow cytometric dot plots and quantitative estimation of polarized and depolarized cells. BA, bafilomycin A1; 3-MA, 3-methyladenine. The data are presented as means±s.e.’s of at least three independent experiments. #P<0.05 vs the control group; $P<0.05 vs Tac group; *P<0.05 vs Tac+3-MA group; @P<0.05 vs Tac+KRGE group; &P<0.05 vs Tac+KRGE+3-MA group.

KRGE reduces Tac-induced impairment of mitochondrial oxygen consumption rate in INS-1 cells. (a) Representative traces of oxygen consumption rate (OCR) measurements performed in INS-1 cells treated with Tac and KRGE with/without 3-MA or BA for 6 h. ATP synthase inhibitor (oligomycin), uncoupler (FCCP), and respiratory chain complex I and III inhibitors (rotenone/antimycin A) were sequentially added. The AUC for the basal OCR (b), ATP-linked OCR (c), maximal OCR (d), and spare respiratory capacity (e) were calculated from the OCR traces. The data are presented as means±s.e.’s of at least three independent experiments. *P<0.05.
Discussion
We designed this study to evaluate whether KRGE is effective in patients with Tac-induced DM, and our study focused on autophagy modulation and oxidative stress in isolated pancreatic islet cells and an experimental model of Tac-induced DM. Results of the present study clearly demonstrate that KRGE is effective in reducing Tac-induced pancreatic islet injury via modulating autophagy, and Tac-induced oxidative stress to mitochondria is causally associated with impaired autophagy. The results of our study provide the rationale for use of KRGE in Tac-induced DM in solid organ transplant recipients.
In vivo and in vitro, we found that Tac increases autophagosome formation (abundant autophagic vacuoles and increased LC3B) and reduces autophagosome clearance (impairment of autophagosome–lysosome fusion and lysosomal acidification), and this was associated with pancreatic islet dysfunction. KRGE modulated this process by decreasing autophagosome formation and improving autophagosome–lysosome fusion and acidification. This improvement was accompanied by improvement of pancreatic islet cell function and reduction of islet cell death. Taken together, Tac-induced DM is a state of excessive autophagosome due to overproduction and impaired clearance of autophagosome. KRGE reduces this burden effectively in pancreatic islet cells, and this may contribute to improvement of pancreatic islet function in TAC-induced DM.
We further evaluated whether the effect of KRGE on Tac-induced pancreatic islet injury is causally related to the autophagy modulation using autophagy inhibitors in cultured INS-1 cells. In Tac-treated beta cells, addition of 3-MA and significantly increased insulin secretion and cell viability, and addition BA decreased both insulin secretion and cell viability. This finding confirms that Tac causes excessive autophagosome formation and lysosomal dysfunction, and altered autophagy process by Tac is causally associated with Tac-induced beta cell dysfunction and cell death. On the other hand, KRGE to Tac+3-MA further increased insulin secretion compared to the Tac+3-MA group and addition of BA to Tac+KRGE diminished insulin secretion. This finding suggests that KRGE has potential of inhibiting autophagosome similar to 3-MA and has ability to improve lysosomal function. These results imply that the KRGE effect is causally associated with autophagy modulation in Tac-induced beta cell injury.
Oxidative stress has an important role in the pathogenesis of Tac-induced pancreatic islet injury.4, 13 Therefore, we evaluated the effect of KRGE on mitochondria, which are susceptible to oxidative damage and are one of the major cellular sources of ROS generation.14, 15 In this study, Tac decreased overall mitochondrial function, demonstrated by increased mitochondrial ROS, superoxide anion production, mitochondrial membrane depolarization, and reduced mitochondrial bioenergetics. However, KRGE decreased or improved those parameters, and this effect was enhanced by 3-MA, but decreased by BA. This finding suggests that the Tac-induced mitochondrial dysfunction is responsible for autophagic cell death, and KRGE decreases autophagic cell death by improving mitochondria function.
Our study has some limitations. First, we did not observe effect of specific ginsenosides of KRGE on autophagy modulation. It is well-known that KRGE contains several ginsenosides (Rg1, Rg3, Rh2, Rb1, and Re), which are known to regulate autophagy.16, 17, 18, 19, 20 Further studies will be needed to define the role of each ginsenoside in Tac-induced DM. Second, we expected the marked effect of KRGE on Tac-induced DM but it did not. The reason for weak effect of KRGE may be related to the use of KRGE rather than specific ginsenoside. Nevertheless, our study reveals scientific evidence that KRGE provide glucose-lowering effect by modulating autophagy in Tac-induced DM.
In conclusion, Tac-induced DM is a state of impaired regulation of autophagy. KRGE modulates autophagy favorably by reducing Tac-induced oxidative stress, and this effect is closely associated with improvement of mitochondrial function.
References
Sharif A . Preventing and managing hyperglycemia in kidney transplant patients. Curr Opin Nephrol Hypertens 2012;21:574–579.
Valderhaug TG, Hjelmesaeth J, Hartmann A et al. The association of early post-transplant glucose levels with long-term mortality. Diabetologia 2011;54:1341–1349.
Tufton N, Ahmad S, Rolfe C et al. New-onset diabetes after renal transplantation. Diabetic Med 2014;31:1284–1292.
Lim SW, Jin L, Jin J et al. Effect of exendin-4 on autophagy clearance in beta cell of rats with tacrolimus-induced diabetes mellitus. Sci Rep 2016;6:29921.
Piao SG, Lim SW, Doh KC et al. Combined treatment of tacrolimus and everolimus increases oxidative stress by pharmacological interactions. Transplantation 2014;98:22–28.
Jin J, Lim SW, Jin L et al. Effects of metformin on hyperglycemia in an experimental model of tacrolimus- and sirolimus-induced diabetic rats. Korean J Intern Med 2017;32:314–322.
Mishra BB, Tiwari VK . Natural products: an evolving role in future drug discovery. Eur J Med Chem 2011;46:4769–4807.
Lim SW, Doh KC, Jin L et al. Oral administration of ginseng ameliorates cyclosporine-induced pancreatic injury in an experimental mouse model. PLoS ONE 2013;8:e72685.
Heo SB, Lim SW, Jhun JY et al. Immunological benefits by ginseng through reciprocal regulation of Th17 and Treg cells during cyclosporine-induced immunosuppression. J Ginseng Res 2016;40:18–27.
Doh KC, Lim SW, Piao SG et al. Ginseng treatment attenuates chronic cyclosporine nephropathy via reducing oxidative stress in an experimental mouse model. Am J Nephrol 2013;37:421–433.
Lim SW, Hyoung BJ, Piao SG et al. Chronic cyclosporine nephropathy is characterized by excessive autophagosome formation and decreased autophagic clearance. Transplantation 2012;94:218–225.
Dott W, Mistry P, Wright J et al. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol 2014;2:224–233.
Jin L, Lim SW, Doh KC et al. Dipeptidyl peptidase IV inhibitor MK-0626 attenuates pancreatic islet injury in tacrolimus-induced diabetic rats. PLoS ONE 2014;9:e100798.
Lin MT, Beal MF . Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–795.
Zorov DB, Filburn CR, Klotz LO et al. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000;192:1001–1014.
Li Y, Wang F, Luo Y . Ginsenoside Rg1 protects against sepsis-associated encephalopathy through beclin 1-independent autophagy in mice. J Surg Res 2017;207:181–189.
Moon JH, Lee JH, Lee YJ et al. Autophagy flux induced by ginsenoside-Rg3 attenuates human prion protein-mediated neurotoxicity and mitochondrial dysfunction. Oncotarget 2016;7:85697–85708.
Yang Z, Zhao T, Liu H et al. Ginsenoside Rh2 inhibits hepatocellular carcinoma through beta-catenin and autophagy. Sci Rep 2016;6:19383.
Chen Z, Lu T, Yue X et al. Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoxicity: with emphasis on autophagy. Neurosci Lett 2010;482:264–268.
Son YM, Kwak CW, Lee YJ et al. Ginsenoside Re enhances survival of human CD4+ T cells through regulation of autophagy. Int Immunopharmacol 2010;10:626–631.
Acknowledgements
We thank Dr Hong-Lim Kim for help with electron microscopy. This work was supported by a grant from the Korean Society of Ginseng funded by the Korea Ginseng Corporation to CWY.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The use of the immunosuppressant drug tacrolimus (tac) is associated with new-onset diabetes after transplantation. In mice with Tac-induced diabetes mellitus, ginseng alleviates islet dysfunction and decreases oxidative stress and autophagic vacuoles. In vitro, ginseng decreases autophagosome formation and attenuates lysosomal degradation, and is accompanied by improved beta cell viability and insulin secretion.
Rights and permissions
About this article

Cite this article
Lim, S., Jin, L., Luo, K. et al. Ginseng extract reduces tacrolimus-induced oxidative stress by modulating autophagy in pancreatic beta cells. Lab Invest 97, 1271–1281 (2017). https://doi.org/10.1038/labinvest.2017.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.75
