
Abstract
Apolipoprotein A1 (ApoA1) is a main protein moiety in high-density lipoprotein (HDL) particles. Generally, ApoA1 and HDL are considered as atheroprotective. In prooxidant and inflammatory microenvironment in the vicinity to the atherosclerotic lesion, ApoA1/HDL are subjected to modification. The chemical modifications such as oxidation, nitration, etc result in altering native architecture of ApoA1 toward dysfunctionality and abnormality. Neutrophil myeloperoxidase has a prominent role in this mechanism. Neo-epitopes could be formed and then exposed that makes them immunogenic. Indeed, these epitopes may be recognized by immune cells and induce production of proatherogenic ApoA1-specific IgG antibodies. These antibodies are biologically relevant because they are able to react with Toll-like receptor (TLR)-2 and TLR4 in target cells and induce a variety of pro-inflammatory responses. Epidemiological and functional studies underline a prognostic value of ApoA1 self-antibodies for several cardiovascular diseases, including myocardial infarction, acute coronary syndrome, and severe carotid stenosis.
Similar content being viewed by others

Main
Atherosclerosis-Associated Inflammation
At present, chronic inflammation is considered as a key pathophysiological mechanism in atherosclerosis.1 Inflammatory reactions influence all studies of atherosclerotic progression from the pro-inflammatory activation of the vascular endothelium until plaque rupture and atherothrombotic events. In brief, pro-inflammatory activated endothelial cells (ECs) resided in atheroprone arterial sites release chemokines and increase surface expression of adhesion molecules. From the blood stream, leukocytes (predominantly monocytes) migrate and attach to the endothelium, and then infiltrate the tunica intima, a subendothelial layer of tunica. In the tunica intima, monocytes differentiate to inflammatory macrophages that acquire capacity to take up native and oxidatively modified LDL. Lipids come to the tunica intima from the arterial lumen across the endothelium. Overload with lipids leads to the transformation of macrophages into foam cells, a hallmark of early atherosclerotic lesions. Vascular smooth muscle cells (VSMCs) also contribute to LDL-induced formation of foam cells.2 Foam cells accumulate in the tunica intima forming the atheroma, a lipid core of the plaque. They also secrete a variety of inflammatory mediators that attract other types of immune cells to the plaque and aggravate inflammation. These mediators also promote hyperplasia of VSMCs that begin to migrate and proliferate, and contribute to the neointimal formation. Inflammatory mediators also facilitate intraplaque neovascularization. The atheroma can partly contibute to the necrotic core formation due to the apoptosis and necrosis of foam cells3 and other cell types (eg, extravasated red blood cells). Hovewer, the necrotic core forms deeply inside the plaque. In advanced plaques, the necrotic core is covered by the fibrous cup composed of neointimal VSMCs and collagen deposits. Unstable lesions are characterized with thin fibrous cap, increased production of inflammatory mediators such as cytokines, matrix metalloproteinases (MMPs), tissue factor, etc, reduction in collagen content, and is often broken. Plaque rupture leads to thrombus formation followed by late atherosclerotic complications such as acute coronary syndrome (ACS), acute myocardial infarction (MI), stroke, and thromboembolism.4
Inflammation has a crucial role on all stages of atherogenesis starting from the transendothelial trafficking of leukocytes in initial steps of atherosclerosis until the plaque rupture. Inflammatory responses regulate overproduction and activity of MMPs involved in pathological arterial wall remodeling and fibrous cup thickening. Inflammatory plaque microenvironment also promotes thrombosis through the induction of hemostatic and procoagulant mechanisms.5
Along with macrophages and other components of innate immunity, adaptive immunity critically influences atherogenic inflammation. In lesions, self-antibodies and various T-cell subsets were found.6 In atherosclerosis, adaptive immunity is driven by a repertoire of plaque T-helper (Th) lymphocytes, which is represented by the pro-inflammatory Th1 cells, anti-inflammatory Th2 cells, and Th17 cells, whose role in atherogenesis is controversial.7, 8 Regulatory T cells are atheroprotective.9 Similarly, the presence of both pro- and anti-inflammatory subpopulations of dendritic cells (DCs)10 and B cells11 is described in the plaque. Indeed, detection of various key autoimmunity-associated mediators such as T cells, B cells, DCs, and self-antibodies may suggest for the occurrence of the autoimmune response in atherosclerosis.
Atherosclerosis-Associated Autoimmunity
In lesions, self-antibodies and T-cell clones specific to different autoantigens were found.5 Patients affected with autoimmune inflammatory diseases such as rheumatoid arthritis (RA), anti-phospholipid syndrome (APS), and systemic lupus erythematosus (SLE) were shown to exhibit higher cardiovascular disease (CVD) risk that is independent on traditional CVD risk factors.12 In CVD patients without autoimmune pathologies, titers of antibodies specific to self-antigens such as oxidized LDL (oxLDL), heat-shock protein (HSP) 60/65, and cardiolipin are increased, and have a prognostic value for prediction of CVD outcomes independently of other CVD risk factors.13 In addition, some self-antibodies (eg, those against HSP60) are able to modulate atherosclerosis progression by binding to Toll-like receptors (TLRs).14 Altogether, these facts support a role of autoimmunity in atherogenesis.
Some investigators proposed that atherosclerosis is an autoimmune disease15, 16 or even has an autoimmune origin.17 The latter is based on the observations suggesting for the existence of immune cross-reactivity between pathogen antigens and host proteins (eg, such as HSP60) due to the sequence similarity.18 In addition, oxidation and other modifications of LDL can generate new epitopes that could be recognized as ‘non-self’ and therefore induce production of self-antibodies.19 However, these oxidation-specific IgM antibodies are the innate immunity components, as they are produced constitutively and do not require T-cell-mediated induction.20 These germline natural IgM antibodies are atheroprotective, as they are involved in the clearance of proatherogenic oxLDL.21
Generation of a new epitope is normally not sufficient enough to induce an antigen-specific immune response because antigen-recognizing DCs should simultaneously receive a second activation signal through TLRs and other pattern recognition receptors (PRRs). In a case of infection, pathogen-associated molecular patterns serve as ligands for PRRs. If infection is absent, PRRs could be stimulated by damage-associated molecular patterns generated by dying and stressed cells, which in turn induces ‘sterile inflammation’.19, 22 Indeed, ‘sterile inflammation’ and molecular mimicry with pathogens may represent the pathways by which self-antibodies specific to atherosclerosis-related antigens can contribute to atherogenesis.1
In contrast to proatherogenic LDL, high-density lipoprotein (HDL) is atheroprotective. Apolipoprotein A1 (apoA1) is a principal protein component of HDL. Plasma HDL cholesterol and ApoA1 levels are associated with lower CVD risk, especially MI risk.23, 24 However, atherosclerosis-related oxidative stress and inflammation lead to oxidation and other modifications of mature ApoA1, a phenomenon that can switch atheroprorective properties of native apoA1 to proatherogenic properties of modified ApoA1.25 ApoA1 modification could induce formation of ApoA1-specific IgG antibodies that exhibit pro-inflammatory properties. In this review, we consider ApoA1 structural and functional properties, and a role of ApoA1-specific antibodies in atherosclerotic disease.
ApoA1 STRUCTURE AND FUNCTION
HDL Particles and ApoA1
Among lipoprotein particles, HDL particles are the smallest (7–12 nm in size), but the densest (1.063–1.21 g/ml) due to the highest ratio between proteins and lipids (50% of the protein component).26 Like other lipoproteins, HDL is mainly produced by the liver. Unlike the larger lipoprotein particles that transport lipids to the cells, HDL participates in removing excessive lipids from the cells. HDL is involved in reverse cholesterol transport (RCT) from macrophages, an atheroprotecive property.27 Other anti-atherosclerotic HDL characteristics involve anti-inflammatory, anti-thrombotic, anti-apoptotic, and vasodilatory effects.28
HDL represents a very heterogeneous assembly of lipids and proteins. In HDL, ~80 proteins is found, the most substantial portion of those (~1/3) is involved in the lipid transfer/metabolism.25 ApoA1 is crucially involved in RCT via the macrophagal ATP-binding cassette transporter ABCA1.29 This apolipoprotein is a main protein constituent of HDL accounting for an ~70% of HDL protein.26 Human mature ApoA1 (molecular weight (m.w.) 28 kDa) comprises 243 amino-acid (a.a.) residues after removing a 24-a.a.-long leader sequence during the post-translational processing. The C-terminal domain of human ApoA1 contains 11 and 22-a.a tandem repeats that compline a total of residues 44–243.30 Each of the repeats has an amphipathic α-helix, which is a major contributor to bind lipids in an exchangeable manner.31 In apolipoproteins, the class A amphipathic helix is a major contributor for binding lipids, which contains a.a. with a positive charge closely to the hydrophobic/hydrophilic surface and a.a. with a negative charge at the nucleus of the polar surface. Class Y and class G* helices are also involved in lipid binding, but have a secondary role. The amphipathic α-helices are critical for interacting with phospholipids and organizing an HDL particle. In ApoA1, the motifs of amphipathic α-helices are evolutionally preserved.32 The total helix content is 53%, although β-sheets contribute for a total of 12% of the a.a. sequence of ApoA1.26
Lipid-Binding and Lipid-Solubilizing Properties of ApoA1
ApoA1 binding to lipids (ie, entrance to a lipid particle) is induced by the interaction between a highly hydrophobic C-terminal domain with the surface lipids of a particle. This contact is followed by formation of α-helices from a random coil in the C-terminal domain. In the lipid particle, the N-terminal helix ApoA1 bundle is then subjected to conformational opening thereby scheduling from hydrophobic helix–helix interactions to helix–lipid interactions.33 Decrease in helix bundle stability facilitates opening and increases interaction with lipids.34 There is no unique mechanism of ApoA1 helix bundle opening within the lipid particle. The opening starts from the separation about a hinge of two helix pairs, but is unclear how the helices are paired.35
In serum phospholipid-coated particles carrying triglyceride- and cholesterol-containing core, ApoA1 incorporation does not stabilize a particle. However, in phospholipid particles, ApoA1 binding may result in particle reorganization and solubilization of the bilayer.26 The suggested mechanism of phospholipid bilayer solubilization involves the insertion of ApoA1 inside the particle followed by the formation of the amphipathic α-helices and interaction with the phospholipid surface. This in turn leads to the fragmentation of the bilayer and formation of discoidal phospholipid particles.36 Rearrangement to discoidal HDL particles is dependent on the concentration of α-helices. When this concentration reaches a critical value, the bilayer begins destabilization and transforms to discoidal particles. The spontaneous solubilization is dependent on the cholesterol content and physical characteristics of the bilayer.37 Typically, discoidal HDL particles contain two or three ApoA1 molecules. Larger particles include more protein.
The C-terminal domain accounts for the almost all ApoA1 lipid-solubilizing properties.38 Interestingly, the isolated C-terminal domain acts as an entire protein, which is able to solubilize phospholipid particles and induce formation of HDL disks of a smaller size.39 Overall, the hydrophobicity of the C-terminal domain and the stability of the N-terminal helix bundle are the major determinants of lipid-solubilizing and lipid-binding properties of ApoA1.
ApoA1 Structure in Discoidal HDL Particles
The organization of a discoidal HDL particle containing two ApoA1 molecules and 140 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine molecules is well characterized and confirmed by different approaches.40 During ApoA1 incorporation to the HDL particle, conformational changes involve a sequence comprising total of 102 a.a. (a.a. 45–53, 66–69, 116–146, and 179–236). These changes lead to an increase in α-helix content by 40%. Almost all ApoA1 molecule is involved in lipid-bound status except for N-terminal six residues and C-terminal seven residues.26 The discoidal HDL particle involves phospholipid layer oriented parallel to the ApoA1 helices that wrap the particle edge like a double belt.41 The elongated helical structure is punctuated by kinks at proline residues.42
In the HDL disk, chemical cross-linking experiments showed that the central helical portion (a.a. 121–142) of one ApoA1 molecule is adjacent to the similar region of its counterpart.40 However, it is unclear whether all of helical regions of both ApoA1 molecules strictly follow this rule43 probably indicating the independent rotation of two ApoA1 antiparallel molecules in the discoidal particle.44 Regarding the N-terminal region, there are data showing that this region can fold back on itself with the degree that depends on the particle size.45, 46
In larger HDL particles (9.6 nm), the helical structure of ApoA1 is well organized. The region involving 125–18 a.a. represents as a disordered loop that either interacts with the phospholipid bilayer or is exposed outside the particle. In contrast, a.a. 159–180 that are involved in the lecithin–cholesterol acyltransferase (LCAT)-activating region of ApoA1 are organized rather as a helix, but not as a random loop.47 In smaller particles (7.8 nm), due to increased packing density of ApoA1, more regions (a.a. 45–57, 115–158, and 190–243) lose the helical structure and become unfolded. These regions correspond to the portions of lipid-free ApoA1 that are mostly non-helical.47 Discoidal HDL particles with the size exceeding 10 nm may include more ApoA1 molecules. In particles with three molecules, at least one should have a helical hairpin conformation.48 In fact, 9.6-nm HDL disks represent HDL particles with the size that provides an option for both ApoA1 molecules to have an optimal conformation.
In HDL disks, amphipathic α-helices of ApoA1 are relatively unstable and can dynamically change the conformation from folded to unfolded47 due to the presence of phospholipids. Indeed, the conformation of lipid-bound ApoA1 is highly flexible. This conformational plasticity in turn promotes remodeling of HDL particles during maturation and metabolism.49
ApoA1 Structure in Spherical HDL Particles
In human blood, circulating HDL are predominantly presented by HDL spheres. Spherical HDL particles contain cholesterol esters and result from free cholesterol-containing discoidal HDL particles after the LCAT action.50 In spherical HDL, ApoA1 molecules envelope ~80% of a total surface thereby underlining the major role of this apolipoprotein in stabilizing HDL structure and shape.34
In the spheres, Silva et al51 showed that ApoA1 maintains the double-belt arrangement such as in discoidal HDL. The trefoil model of ApoA1 conformation was proposed, where a half of each of three ApoA1 molecules is flexed by 60° to maintain a paired double-belt arrangement between adjacent protein segments.51 In the full-length protein, the flexion site was supposed to occur close to a.a. 65 and 185.45 The trefoil model is not a unique pattern of ApoA1 arrangement in HDL spheres. A helical dimer/hairpin model was proposed, with two molecules forming the intertwined helical dimeric structure and the third, which forms a helical hairpin.52 Both ApoA1 conformations could putatively co-present in the HDL spheroid particle due to the high flexible ApoA1 structure.
Like in discoidal particles, the diameter of an HDL sphere also influences the ApoA1 helical structure. In spheres whose size is close to 10 nm, the molecular organization of ApoA1 is very similar to the ApoA1 structure in discoidal particles. Reduction in the spherical particle size increases the protein-packing density that in turn leads to a less folded structural organization.51, 53 HDL size also influences the conformation of all ApoA1 structural segments, with the central region (a.a. 121–165), whose conformation is especially sensitive to the particle size.54
A dynamic ApoA1 conformation involves a possibility of ApoA1 reversible dissociation/association with the HDL particle.55 During dissociation, the ApoA1 molecule is anchored on the particle through the C-terminal domain, with the N-terminal domain in closed conformation outside of the particle. Indeed, an exchangeable pool of ApoA1 exists in the plasma that involves lipid-bound and lipid-free (also called pre-β1-HDL) molecules.56
Lipid-free ApoA1 is a minor fraction of serum ApoA1, which accounts for 5% of a total ApoA1.57 It should be mentioned that most of immunological assays quantifying anti-apoA-1 IgG titers were developed using delipidated ApoA1. As lipidation of ApoA1 induces conformational changes in the ApoA1 molecule, using these assays for measuring anti-apoA-1 IgG levels may potentially lead to biased results.
Involvement of ApoA1 in Cholesterol Esterification
Remodeling of HDL particles by plasma factors is an essential step in HDL biogenesis. As mentioned above, rearrangements in HDL particles frequently lead to exchangeable dissociation of ApoA1 from the HDL particle into the aqueous phase. The reversible exchange between lipid-free and lipid-bound ApoA1 is crucial for HDL metabolism and remodeling.58
In plasma, ApoA1 is a key stimulator for LCAT, an enzyme secreted by the liver. In serum lipoproteins, LCAT is critically involved in esterification of unesterified (free) cholesterol.59 This enzyme has 416 a.a. and represents a glycoprotein with m.w. of ~60 kDa. Ser 181 is located in the active site and is crucial for the LCAT catalytic activity.60 In the ApoA1 molecule, a.a.143–187 are involved in LCAT binding to the HDL particle. In the LCAT-binding site, arginine residues (ie, Arg 149, Arg 153, and Arg 160) are crucially contributed to the interaction with the enzyme.61
The enzyme has a high affinity of binding to HDL disks,62 where LCAT performs synthesis of cholesterol esters. In contrast to free cholesterol, cholesterol esters are badly soluble in the phospholipid bilayer and form a lipid core in the HDL particle. This leads to the transformation of discoidal HDL to spherical HDL.50 Indeed, genetic deletion of LCAT in mice results in lowered concentrations of HDL spheres, although discoidal HDL levels are elevated.63
Involvement of ApoA1 in RCT
ApoA1 regulates RCT through interaction with ABCA1. ABCA1 is a 2261-a.a.-long membrane transporter that mediates transfer of cholesterol and phospholipids from the cells. ApoA1 cooperates with ABCA1 in the generation of discoidal nascent HDL particles.64 The ability of ApoA1 to solubilize and bind lipids has a key role in formation of nascent HDL.65
ApoA1/ABCA1-dependent generation of nascent HDL particles involves three stages. On the first stage, ApoA1 (lipid free or lipid bound) binds to ABCA1 that results in enhancing activity of the transporter in translocating phospholipids from the inner plasma membrane layer to the outer layer. Lipid accumulation leads to the membrane bulging and formation of microdomains enriched with free cholesterol and phospholipids (stage 2). On the third stage, ApoA1 binds to these membrane microdomains and induces their solubilization with formation of discoidal nascent HDL.66 The amphipathic α-helices of apoA-I are critically involved in this mechanism.67 Nascent HDL particles (7–17 nm in diameter) are originally discoidal and include 2–4 ApoA1 molecules.68 In a case of high phospholipid content, generation of larger HDL particles is enhanced.69, 70
As ApoA1 is directly involved in lipid metabolism, this property is important for considering this apolipoprotein as one of key molecular players in the pathogenesis of cardiometabolic diseases, such as atherosclerosis.
ROLE OF MODIFIED ApoA1 IN ATHEROSCLEROSIS
Generally, ApoA1/HDL fraction of serum lipoproteins is atheroprotective, as it is involved in removal of excessive cholesterol and phospholipids from ABCA1-expressing cells such as macrophages and VSMCs thereby preventing formation of foam cells, which are key contributors to the atheroma formation.71 In macrophages and VSMCs derived from atherosclerotic plaques, ABCA1 expression and ApoA1-binding capacity is markedly reduced that in turn diminishes ApoA1-mediated cholesterol efflux and promotes lipid deposition.71, 72, 73, 74
Notably, as mentioned above, ApoA1 could be modified in atherosclerotic plaques through oxidation, nitration, and other mechanisms. This could greatly impair functional properties of intact ApoA1 and even make it dysfunctional. In the lesions, multiple modified ApoA1 variants were found. For example, ApoA1 nitrated at Tyr 166 is abundant (~8% of total ApoA1) in human lesions. In serum, this variant is existed in lipid-poor state due to a low lipid-binding capacity and 10-fold reduced ability to bind LCAT.75
ApoA1 oxidation and chlotorination is mainly mediated by neutrophil myeloperoxidase (MPO).76 MPO-mediated oxidation of Trp 72 leads to the formation of the oxTrp72-ApoA1 variant that is abundantly present (20% of total apoA1 in atherosclerotic vessels) in the plaque and lack an ability to activate ABCA1.77 MPO-dependent nitration and chlorination of ApoA1 at Tyr 192 along with methionine oxidation results in a loss of ability to activate RCT.78, 79
Modified ApoA1 form HDL with impaired function. For example, lipid-free ApoA1 treated with hypothiocyanous acid (HOSCN), a major oxidant generated by MPO, forms HDL that fails capacity to regulate cholesterol efflux, but remains anti-inflammatory properties. ApoA1 exposed to cyanate (HOCN), a product of HOSCN decomposition, lack the ability to control RCT and become pro-inflammatory.80, 81 Compared with native HDL, in ApoE-deficient mice, MPO-modified HDL lose anti-atherogenic properties as they unable to stabilize plaque, prevent foam cell formation, and suppress formation of macrophages with the pro-inflammatory phenotype.34 MPO-modified HDL cannot bind to the HDL receptor and scavenger receptor B1, and promote pro-inflammatory activities such as upregulation of nuclear factor (NF)-κB and expression of adhesion molecules by ECs.82
Furthermore, both modified HDL and ApoA1 become immunogenic because modification creates new epitopes that could be recognized by immune cells and induce humoral immune response and anti-ApoA1 antibody production.
ApoA1 EPITOPES
Immunogenic ApoA1 epitopes are highly heterogeneous. They include linear and conformational (continuous and discontinuous) structures, neo-epitopes, and mimo-epitopes. To date, many immunogenic epitopes specific for lipid-free and lipid-bound human ApoA1 were identified. This resulted in the development of monoclonal antibodies specific to these epitopes. In modified ApoA1, such epitopes were also found. For example, in MPO-modified ApoA1 found in human plaques has oxindolyl alanine (2-OH-Trp) resulted from the oxidation of Trp 72. The 2-OH-Trp moiety is the immunogenic epitope recognized by a high-affinity monoclonal antibody that is also capable to bind to HDL treated with MPO.77 Recently, Teixeira et al83 found two novel immunoreactive peptides recognizable by self-antibodies from MI patients with higher titers of ApoA1 IgG. The first peptide (peptide C) comprises 28 residues (Gln240–Gln267) and includes a whole non-polar α-helix involved in lipid solubilization. The second peptide represents pyroglutamic variant of peptide C derived from modified ApoA1 and containing cyclization of the free amino group of glutamine.83
ANTI-ApoA1 SELF-ANTIBODIES IN AUTOIMMUNE PATHOLOGIES
Anti-ApoA1 IgG autoantibodies were first detected in patients affected with autoimmune disorders such as SLE and primary APS. Merill et al84 found antibodies reactive against human ApoA1 in a phage display library constructed on the basis of sera from SLE subjects antibodies reactive against human ApoA1. Increased titers of these antibodies were then observed in SLE individuals (32.5%) and primary SLE subjects (22.9%).85 The occurrence of ApoA1 antibodies was associated with the presence of anti-β2-glycoprotein (β2GPI) antibodies and reacted with the highest affinity with mature HDL.86 Analysis of six ApoA1 antibodies isolated from two SLE patients showed their low specificity because the antibodies were cross-reactive with thrombin, cardiolipin, and single-strand DNA.87 Further studies showed correlation between levels of ApoA1 antibodies and disease severity, and carotid intima-media thickness in SLE patients.88, 89, 90
Recently, in a longitudinal study, Croca et al91 also reported significant association between anti-ApoA1 IgG levels and SLE disease activity, but failed to show correlation between early anti-apoA-1 IgG positivity and morbidity, mortality, and CVD risk, which suggests about a lack of prognostic value of anti-ApoA1 antibodies for SLE-induced death or cardiovascular complications in SLE.
Vuilleumier et al92 then found association between ApoA1 antibodies and RA observing higher levels of antibodies in affected individuals compared with matched controls (17 vs 2%). Titers of ApoA1 antibodies are also correlated with increased levels of oxLDL and showed association with clinically manifested CVD. In a follow-up study, Finkh et al93 found that anti-ApoA1 antibodies can serve as an independent predictor of CVD risk in RA subjects and be associated with increased production of pro-inflammatory cytokines.
PROATHEROGENIC FUNCTIONAL EFFECTS OF ANTI-ApoA1 SELF-ANTIBODIES IN CVD
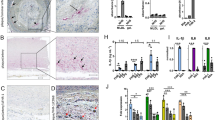
In cardiovascular disorders, anti-ApoA1 autoantibodies are functionally important (Figure 1a–c). As mentioned above, anti-ApoA1 autoantibodies belong to G class and possess pro-inflammatory properties.94 A first evidence highlighting a possible link between ApoA1 IgG antibodies titers and atherosclerosis was obtained by Delgado Alves et al95 who observed a reverse correlation between anti-HDL IgG and paraoxonase-1 (PON-1) activity (an enzyme responsible for antioxidant properties of HDL), and with the total antioxidant capacitance of the sera of SLE patients with atherosclerosis. In fact, this correlation can be explained by the involvement of anti-ApoA1 antibodies in the impairment of HDL function.96, 97 Similarly, in experimental murine lupus model, elevated titers of ApoA1 antibodies were shown to correlate with reduced PON-1 activity and decreased HDL cholesterol concentrations.98 This in turn reflects the pathological role of ApoA1 IgG in atherosclerosis by disrupting anti-inflammatory and antioxidant properties of HDL mediated by PON-1, an HDL-associated esterase (Figure 1c). In ApoE-deficient mice, immunization with ApoA1 antibodies led to plaque expansion and destabilization suggesting for atherogenic effects.99

ApoA1-specific self-antibodies and their functional effects in cardiovascular pathophysiology (a-c). (a) Proatherogenic effects of ApoA1-specific self-antibodies in vascular cells. The antibodies are capable to bind to Toll-like receptor (TLR)-2 and TLR4. As a result, TLR2/TLR4/CD14 mechanism mediates signaling that leads to the activation of a transcription nuclear factor (NF)-κB, which in turn directs expression of inflammatory messengers such as tumor necrosis factor (TNF)α, interleukin (IL)-6, IL-8, chemokine (C–C motif) ligand 2 (CCL2), and matrix metalloproteinase (MMP-9). This effect could probably deliver initiation or prolongation of inflammation in arteries. (b) Effects of ApoA1-specific self-antibodies on cardiac muscle cells. Normally, aldosterone alone is able to increase myocardial beating through binding to the mineralocorticoid receptor and further signaling via phosphatidylinositol-3-kinase (PI3K)/Akt pathway. That causes opening of L-type Ca2+ channels, which thus activates potassium/calcium exchangers and leads to the depolarization of the plasma membrane of cardiomyocytes. Indeed, that stimulates heart beating. ApoA1 antibodies can mimic that through interaction with TLR2/4 mediated by CD14. Whether TLR2/4 transduction mediates the ApoA1 antibody-dependent pathway to activate protein kinase A (PKA) is unclear. However, the antibodies via binding to TLR2/TLR4/CD14 could provide alternative (or concomitant) signal to PI3K with subsequent stimulation of Src-dependent pathway, that finally activates L-type Ca2+ channel. (c) Anti-ApoA1 antibodies cause HDL dysfunction. In an HDL particle, PON-1 can interact with MPO. This interaction leads to partial MPO inhibition. Reciprocally, MPO inhibits PON-1 activity through oxidation at tyrosine 71, which is crucial for HDL binding and PON-1 function. Indeed, balancing between PON-1 and MPO activity has a key role in establishing antioxidant and anti-inflammatory properties of HDL. Anti-ApoA1 antibodies decrease PON-1 activity, which leads to PON-1/MPO imbalance and MPO activation. MPO is involved in oxidative modification of lipids, including generation of proatherogenic oxLDL from LDL. Increased oxLDL levels display a wide spectrum of proatherogenic effects listed in the figure. ApoA1, apolipoprotein A1; HDL, high-density lipoprotein; MPO, myeloperoxidase; PON-1, paraoxonase-1; oxLDL, oxidized LDL.
In vitro experiments showed that ApoA1 self-antibodies are functionally active. These antibodies are capable to bind to TLR2 and influence intracellular signaling in a TLR2/CD14-dependent manner.100 The ability of ApoA1 self-antibodies to recognize TLR2 arises from the partial homology between TLR2 and ApoA1 a.a. sequences that therefore induces cross-reactivity.100 Indeed, the presence of a partial homology between TLR2 and ApoA1 a.a. sequences presumes the possibility that anti-apoA-1 IgG response can take place from a pathogen exposure cannot be excluded. In addition, antibody-dependent stimulation of TLR2 leads to secretion of inflammatory mediators such as IL (interleukin)-6, IL-8, tumor necrosis factor (TNF)-α, MMP-9, and chemokine (C–C motif) ligand 2 by macrophages derived from MI patients.100 Induction of expression of these mediators is directed by NF-kB, a pro-inflammatory transcription factor (Figure 1a). Recently, Montecucco et al101 showed that TLR4 could be also involved in proatherogenic effects of ApoA1 antibodies in ApoE-deficient mice. ApoA1 antibodies were also shown to enhance migration of neutrophils toward gradient of chemokine (C–X motif) ligand 8.99
A correlation between serum concentrations of ApoA1 antibodies and resting heart rate (RHR) was observed.102 RHR serves as an established prognostic indicator of CVD progression after MI.103, 104 In cultured rat cardiomyocytes treated with aldosterone and ApoA1 antibodies, contraction rate was found to progressively correlate with titers of ApoA1 antibodies in a dose-dependent manner.102 The positive chronotropic effect is mediated through upregulation of L-type Ca2+ channels via phosphatidyl 3-kinase (PI3K)- and protein kinase A-dependent mechanisms.105 In cardiac muscle cells, aldosterone activates L-type Ca2+ channels through binding to the mineralocorticoid receptor followed by upregulation of the PI3K/Akt pathway, although the TLR2/TLR4/CD14 complex mediates antibody-dependent stimulation of PI3K and Src (Figure 1b).106
Further functional studies are required to explain multiple correlations observed between titers of circulating ApoA1 antibodies and various CVD risk parameters that will be considered in the next section. Immunohistochemical studies showed that there is a continuous increase of volume of ApoA1 during the development of atherosclerotic lesions, and that atherosclerotic plaques contain ApoA1 localized as intracellularly and extracellularly (Figure 2).107 Despite a large volume of ApoA1 in atherosclerotic plaques, ApoA1 recovered from human atheroma are dysfunctional as result of an extensive oxidization by MPO.77

ApoA1 in a human atherosclerotic plaque of the human aorta. Large arrows show the extracellular location of Apo1 in the intimal matrix, whereas small arrows show the presence of Apo1 intracellularly, predominantly in intimal cells that exhibit a foam cell appearance. Identification of Apo1 with the use of anti-ApoA1 antibody (Abcam; cat# ab7613); Immunoperoxidase technique; counterstain with hematoxylin; and magnification: × 400. ApoA1, apolipoprotein A1.
Titers of anti-ApoA1 antibodies were shown to be associated with characteristics that increase cardiovascular risk in subgroups of patients affected with disorders that themselves can serve as independent cardiovascular risk factors or linked with atherosclerosis. In patients with periodontitis, the antibodies levels showed association with pathological ankle-brachial index (a parameter to predict the severity of peripheral arterial disease) in subjects younger than 50 years.108 The titers of ApoA1 antibodies correlated with dialysis vintage, a major determinant of vascular calcification and cardiovascular risk in patients with kidney failure.109 Finally, type 2 diabetic patients who were positive for ApoA1 antibodies also had significantly higher cardiovascular risk compared with ApoA1 IgG-negative diabetic subjects thereby indicating that anti-apoA-1 IgG is a cardiovascular risk biomarker in diabetic patients.110
The presence of anti-HDL antibodies was recently reported.111, 112 However, it is unknown whether anti-HDL antibodies are similar to anti-ApoA1 IgG or represent a different entity. A chance that anti-HDL antibodies are different from anti-ApoA1 IgG is likely to be as delipidated ApoA1 form exists in the circulation. Compared with anti-ApoA1 IgG, anti-HDL antibodies may not recognize this form because ApoA1 is heavily lipidated in an HDL particle.
Anti-ApoA1 SELF-ANTIBODIES AS PROGNOSTIC AND DIAGNOSTIC MARKERS FOR CVD
The diagnostic and prognostic value of ApoA1 self-antibodies in cardiovascular pathology was evaluated in patients affected with various CVD mostly with MI (data are summarized in Table 1). Overall, increased titers of ApoA1 antibodies were shown to correlate with increased levels of inflammatory markers in MI patients100 and with higher oxLDL in ASC subjects.113 In patients with severe carotid stenosis, elevated levels of ApoA1 antibodies directly correlated with various pro-inflammatory signatures such as counts of intraplaque macrophages and neutrophils, and MMP-9 levels, but inversely correlated with a carotid plaque collagen content thereby suggesting for a plaque-destabilizing role of the antibodies.99 These data clearly reflect proatherogenic and pro-inflammatory effects ApoA1 antibodies.
Noteworthy, prospective cohort studies showed a prognostic value of ApoA1 antibodies as a predictor of unfavorable cardiovascular events in patients with MI,102, 114 severe carotid stenosis (in patients, who underwent carotid endarterectomy),115, 116 and RA.117, 118 However, all these data were obtained in a single-center (ie, based on the Geneva University Hospital) cohort studies. In a multicenter study involved a total of 1072 subjects for suspected non-ST segment elevation acute MI (NSTEMI), ApoA1 antibodies failed to be a diagnostic and prognostic marker for NSTEMI.119 Obviously, it would be beneficial to reproduce these correlations in independent cohorts of patients.
Compared with other circulating self-antibodies tested such as those against cardiolipin, HSP60, phosphorylcholine, and glycophosphatidylinositol, ApoA1 self-antibodies are superior as predictive markers for adverse cardiovascular events in MI.120 Similarly, in RA patients, titers of ApoA1 antibodies were found to be the best predictors of future adverse cardiovascular events as compared with oxLDL and pro-brain natriuretic peptide.118 In summary, elevated titers of ApoA1 antibodies are associated with a worse cardiovascular prognosis. Probably, ApoA1 antibodies could serve as a promising marker of cardiovascular autoimmunity.121, 122
CONCLUDING REMARKS
Function of intact ApoA1 in normal and diseased vessels is established as cardioprotective. When modified, ApoA1 goes to the opposite role being proatherogenic and pro-inflammatory moiety that in turn might be as a source of immune response due to the formation of neo-epitopes in part. However, as this possibility was not shown experimentally, it still represents a possible hypothesis. It is unclear whether lipid-free or lipid-bound ApoA1 is the superior target for modification. That is difficult to measure because of the flexibility of ApoA1 within the lipid particle and its ability to lipid solubilization.
The proatherogic properties of ApoA1 self-antibodies are presented. The titers of antibodies are associated with proatherogenic characteristics in MI, carotid stenosis, and RA subjects (those have no clinical CVD). However, this was shown in single-center studies. The only single multicenter study published so far included only NSTEMI patients and did not confirm the prognostic value of anti-ApoA1 antibodies for CVD reported in single-center studies. Therefore, further large multicenter studies are required to draw definite conclusions regarding the possible cardiovascular prognostic role of anti-apoA1 IgG.
Hopefully, ApoA1 antibodies are perspective to be used as an autoimmune biomarker of CVD. Knowing whether anti-apoA1 IgG could provide higher prognostic value than other autoantibodies deserves further investigation.
References
Hansson GK, Nilsson J . Introduction: atherosclerosis as inflammation: a controversial concept becomes accepted. J Intern Med 2008;263:462–463.
Costales P, Fuentes-Prior P, Castellano J et al. K domain CR9 of low density lipoprotein (LDL) receptor-related protein 1 (LRP1) is critical for aggregated LDL-induced foam cell formation from human vascular smooth muscle cells. J Biol Chem 2015;290:14852–14865.
Hegyi L, Skepper JN, Cary NR et al. Foam cell apoptosis and the development of the lipid core of human atherosclerosis. J Pathol 1996;180:423–429.
Badimon L, Vilahur G . Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med 2014;276:618–632.
Hansson GK, Libby P, Tabas I . Inflammation and plaque vulnerability. J Intern Med 2015;278:483–493.
Hansson GK, Holm J, Jonasson L . Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol 1989;135:169–175.
Andersson J, Libby P, Hansson GK . Adaptive immunity and atherosclerosis. Clin Immunol 2010;134:33–46.
Taleb S, Tedgui A, Mallat Z . IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb Vasc Biol 2015;35:258–264.
Chistiakov DA, Bobryshev YV, Orekhov AN . Heterogeneity of Tregs and the complexity in the IL-12 cytokine family signaling in driving T-cell immune responses in atherosclerotic vessels. Mol Immunol 2015;65:133–138.
Chistiakov DA, Sobenin IA, Orekhov AN et al. Dendritic cells in atherosclerotic inflammation: the complexity of functions and the peculiarities of pathophysiological effects. Front Physiol 2014;5:196.
Perry HM, McNamara CA . Refining the role of B cells in atherosclerosis. Arterioscler Thromb Vasc Biol 2012;32:1548–1549.
Schuett KA, Lehrke M, Marx N et al. High-risk cardiovascular patients: clinical features, comorbidities, and interconnecting mechanisms. Front Immunol 2015;6:591.
Roux-Lombard P, Pagano S, Montecucco F et al. Auto-antibodies as emergent prognostic markers and possible mediators of ischemic cardiovascular diseases. Clin Rev Allergy Immunol 2013;44:84–97.
Carbone F, Nencioni A, Mach F et al. Evidence on the pathogenic role of auto-antibodies in acute cardiovascular diseases. Thromb Haemost 2013;109:854–868.
Blasi C . The autoimmune origin of atherosclerosis. Atherosclerosis 2008;201:17–32.
Pereira IA, Borba EF . The role of inflammation, humoral and cell mediated autoimmunity in the pathogenesis of atherosclerosis. Swiss Med Wkly 2008;138:534–539.
Grundtman C, Wick G . The autoimmune concept of atherosclerosis. Curr Opin Lipidol 2011;22:327–334.
Grundtman C, Kreutmayer SB, Almanzar G et al. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arterioscler Thromb Vasc Biol 2011;31:960–968.
Miller YI, Choi SH, Wiesner P et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res 2011;108:235–248.
Chou MY, Hartvigsen K, Hansen LF et al. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med 2008;263:479–488.
Binder CJ . Natural IgM antibodies against oxidation-specific epitopes. J Clin Immunol 2010;30 (Suppl 1):S56–S60.
Yokota S, Minota S, Fujii N . Anti-HSP auto-antibodies enhance HSP-induced pro-inflammatory cytokine production in human monocytic cells via Toll-like receptors. Int Immunol 2006;18:573–580.
Karthikeyan G, Teo KK, Islam S et al. Lipid profile, plasma apolipoproteins, and risk of a first myocardial infarction among Asians: an analysis from the INTERHEART Study. J Am Coll Cardiol 2009;53:244–253.
Shioji K, Mannami T, Kokubo Y et al. An association analysis between ApoA1 polymorphisms and the high-density lipoprotein (HDL) cholesterol level and myocardial infarction (MI) in Japanese. J Hum Genet 2004;49:433–439.
Navab M, Reddy ST, Van Lenten BJ et al. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol 2011;8:222–232.
Phillips MC . New insights into the determination of HDL structure by apolipoproteins: thematic review series: high density lipoprotein structure, function, and metabolism. J Lipid Res 2013;54:2034–2048.
Lund-Katz S, Phillips MC . High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem 2010;51:183–227.
Besler C, Lüscher TF, Landmesser U . Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med 2012;4:251–268.
Mukhamedova N, Escher G, D'Souza W et al. Enhancing apolipoprotein A-I-dependent cholesterol efflux elevates cholesterol export from macrophages in vivo. J Lipid Res 2008;49:2312–2322.
Li WH, Tanimura M, Luo CC et al. The apolipoprotein multigene family: biosynthesis, structure, structure-function relationships, and evolution. J Lipid Res 1988;29:245–271.
Segrest JP, Jones MK, De Loof H et al. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res 1992;33:141–166.
Bashtovyy D, Jones MK, Anantharamaiah GM et al. Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J Lipid Res 2011;52:435–450.
Saito H, Dhanasekaran P, Nguyen D et al. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J Biol Chem 2003;278:23227–23232.
Hewing B, Parathath S, Barrett T et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2014;34:779–789.
Narayanaswami V, Kiss RS, Weers PM . The helix bundle: a reversible lipid binding motif. Comp Biochem Physiol A Mol Integr Physiol 2010;155:123–133.
Lund-Katz S, Nguyen D, Dhanasekaran P et al. Surface plasmon resonance analysis of the mechanism of binding of apoA-I to high density lipoprotein particles. J Lipid Res 2010;51:606–617.
Fukuda M, Nakano M, Sriwongsitanont S et al. Spontaneous reconstitution of discoidal HDL from sphingomyelin-containing model membranes by apolipoprotein A-I. J Lipid Res 2007;48:882–889.
Tanaka M, Koyama M, Dhanasekaran P et al. Influence of tertiary structure domain properties on the functionality of apolipoprotein A-I. Biochemistry 2008;47:2172–2180.
Lyssenko NN, Hata M, Dhanasekaran P et al. Influence of C-terminal α-helix hydrophobicity and aromatic amino acid content on apolipoprotein A-I functionality. Biochim Biophys Acta 2012;1821:456–463.
Davidson WS, Thompson TB . The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem 2007;282:22249–22253.
Koppaka V, Silvestro L, Engler JA et al. The structure of human lipoprotein A-I. Evidence for the "belt" model. J Biol Chem 1999;274:14541–14544.
Segrest JP, Jones MK, Klon AE et al. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem 1999;274:31755–31758.
Li L, Li S, Jones MK et al. Rotational and hinge dynamics of discoidal high density lipoproteins probed by interchain disulfide bond formation. Biochim Biophys Acta 2012;1821:481–489.
Caulfield TR . Inter-ring rotation of apolipoprotein A-I protein monomers for the double-belt model using biased molecular dynamics. J Mol Graph Model 2011;29:1006–1014.
Gursky O . Crystal structure of Δ(185-243)ApoA-I suggests a mechanistic framework for the protein adaptation to the changing lipid load in good cholesterol: from flatland to sphereland via double belt, belt buckle, double hairpin and trefoil/tetrafoil. J Mol Biol 2013;425:1–16.
Mei X, Atkinson D . Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem 2015;286:38570–38582.
Sevugan Chetty P, Mayne L, Kan ZY et al. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA 2012;109:11687–11692.
Davidson WS, Silva RA . Apolipoprotein structural organization in high density lipoproteins: belts, bundles, hinges and hairpins. Curr Opin Lipidol 2005;16:295–300.
Zannis VI, Fotakis P, Koukos G et al. HDL biogenesis, remodeling, and catabolism. Handb Exp Pharmacol 2015;224:53–111.
Ferretti G, Bacchetti T, Nègre-Salvayre A et al. Structural modifications of HDL and functional consequences. Atherosclerosis 2006;184:1–7.
Silva RA, Huang R, Morris J et al. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci USA 2008;105:12176–12181.
Wu Z, Gogonea V, Lee X et al. The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. J Biol Chem 2011;286:12495–12508.
Huang R, Silva RA, Jerome WG et al. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol 2011;18:416–422.
Curtiss LK, Bonnet DJ, Rye KA . The conformation of apolipoprotein A-I in high-density lipoproteins is influenced by core lipid composition and particle size: a surface plasmon resonance study. Biochemistry 2000;39:5712–5721.
Cavigiolio G, Geier EG, Shao B et al. Exchange of apolipoprotein A-I between lipid-associated and lipid-free states: a potential target for oxidative generation of dysfunctional high density lipoproteins. J Biol Chem 2010;285:18847–18857.
Kane JP, Malloy MJ . Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol 2012;23:367–371.
von Eckardstein A, Huang Y, Assmann G . Physiological role and clinical relevance of high-density lipoprotein subclasses. Curr Opin Lipidol 1994;5:404–416.
Pownall HJ, Ehnholm C . The unique role of apolipoprotein A-I in HDL remodeling and metabolism. Curr Opin Lipidol 2006;17:209–213.
Zannis VI, Chroni A, Krieger M . Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med (Berl) 2006;84:276–294.
Jonas A . Lecithin cholesterol acyltransferase. Biochim Biophys Acta 2000;1529:245–256.
Roosbeek S, Vanloo B, Duverger N et al. Three arginine residues in apolipoprotein A-I are critical for activation of lecithin:cholesterol acyltransferase. J Lipid Res 2001;42:31–40.
Dergunov AD . Kinetic analysis of lecithin: cholesterol acyltransferase activity toward discoidal HDL. Lipids 2011;46:1075–1079.
Santamarina-Fojo S, Lambert G, Hoeg JM et al. Lecithin-cholesterol acyltransferase: role in lipoprotein metabolism, reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol 2000;11:267–275.
Wang S, Smith JD . ABCA1 and nascent HDL biogenesis. Biofactors 2014;40:547–554.
Vedhachalam C, Duong PT, Nickel M et al. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem 2007;282:25123–25130.
Vedhachalam C, Chetty PS, Nickel M et al. Influence of apolipoprotein (Apo) A-I structure on nascent high density lipoprotein (HDL) particle size distribution. J Biol Chem 2010;285:31965–31973.
Vedhachalam C, Liu L, Nickel M et al. Influence of ApoA-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem 2004;279:49931–49939.
Duong PT, Collins HL, Nickel M et al. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res 2006;47:832–843.
Lyssenko NN, Brubaker G, Smith BD et al. A novel compound inhibits reconstituted high-density lipoprotein assembly and blocks nascent high-density lipoprotein biogenesis downstream of apolipoprotein AI binding to ATP-binding cassette transporter A1-expressing cells. Arterioscler Thromb Vasc Biol 2011;31:2700–2706.
Lyssenko NN, Nickel M, Tang C et al. Factors controlling nascent high-density lipoprotein particle heterogeneity: ATP-binding cassette transporter A1 activity and cell lipid and apolipoprotein AI availability. FASEB J 2013;27:2880–2892.
Allahverdian S, Pannu PS, Francis GA . Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc Res 2012;95:165–172.
Attie AD, Kastelein JP, Hayden MR . Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res 2001;42:1717–1726.
Choi HY, Rahmani M, Wong BW et al. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation 2009;119:3223–3231.
Yu XH, Fu YC, Zhang DW et al. Foam cells in atherosclerosis. Clin Chim Acta 2013;424:245–252.
DiDonato JA, Aulak K, Huang Y et al. Site-specific nitration of apolipoprotein A-I at tyrosine 166 is both abundant within human atherosclerotic plaque and dysfunctional. J Biol Chem 2014;289:10276–10292.
Zheng L, Nukuna B, Brennan ML et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest 2004;114:529–541.
Huang Y, DiDonato JA, Levison BS et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med 2014;20:193–203.
Shao B, Bergt C, Fu X et al. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J Biol Chem 2005;280:5983–5993.
Shao B, Oda MN, Bergt C et al. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem 2006;281:9001–9004.
Hadfield KA, Pattison DI, Brown BE et al. Myeloperoxidase-derived oxidants modify apolipoprotein A-I and generate dysfunctional high-density lipoproteins: comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCl). Biochem J 2013;449:531–542.
Rosenson RS, Brewer HB Jr, Ansell BJ et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol 2016;13:48–60.
Undurti A, Huang Y, Lupica JA et al. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem 2009;284:30825–30835.
Teixeira PC, Ducret A, Ferber P et al. Definition of human apolipoprotein A-I epitopes recognized by autoantibodies present in patients with cardiovascular diseases. J Biol Chem 2014;289:28249–28259.
Merrill JT, Rivkin E, Shen C et al. Selection of a gene for apolipoprotein A1 using autoantibodies from a patient with systemic lupus erythematosus. Arthritis Rheum 1995;38:1655–1659.
Dinu AR, Merrill JT, Shen C et al. Frequency of antibodies to the cholesterol transport protein apolipoprotein A1 in patients with SLE. Lupus 1998;7:355–360.
Delgado Alves J, Kumar S, Isenberg DA . Cross-reactivity between anti-cardiolipin, anti-high-density lipoprotein and anti-apolipoprotein A-I IgG antibodies in patients with systemic lupus erythematosus and primary antiphospholipid syndrome. Rheumatology (Oxford) 2003;42:893–899.
Abe H, Tsuboi N, Suzuki S et al. Anti-apolipoprotein A-I autoantibody: characterization of monoclonal autoantibodies from patients with systemic lupus erythematosus. J Rheumatol 2001;28:990–995.
Shoenfeld Y, Szyper-Kravitz M, Witte T et al. Autoantibodies against protective molecules—C1q, C-reactive protein, serum amyloid P, mannose-binding lectin, and apolipoprotein A1: prevalence in systemic lupus erythematosus. Ann NY Acad Sci 2007;1108:227–239.
O'Neill SG, Giles I, Lambrianides A et al. Antibodies to apolipoprotein A-I, high-density lipoprotein, and C-reactive protein are associated with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum 2010;62:845–854.
Radwan MM, El-Lebedy D, Fouda R et al. Anti-apolipoprotein A-1 antibodies and carotid intima-media thickness in Egyptian women with systemic lupus erythematosus. Clin Rheumatol 2014;33:493–498.
Croca S, Bassett P, Chambers S et al. IgG anti-apolipoprotein A-1 antibodies in patients with systemic lupus erythematosus are associated with disease activity and corticosteroid therapy: an observational study. Arthritis Res Ther 2015;17:26.
Vuilleumier N, Bratt J, Alizadeh R et al. Anti-apoA-1 IgG and oxidized LDL are raised in rheumatoid arthritis (RA): potential associations with cardiovascular disease and RA disease activity. Scand J Rheumatol 2010;39:447–453.
Finckh A, Courvoisier DS, Pagano S et al. Evaluation of cardiovascular risk in patients with rheumatoid arthritis: do cardiovascular biomarkers offer added predictive ability over established clinical risk scores? Arthritis Care Res (Hoboken) 2012;64:817–825.
Tanaka M, Dhanasekaran P, Nguyen D et al. Influence of N-terminal helix bundle stability on the lipid-binding properties of human apolipoprotein A-I. Biochim Biophys Acta 2011;1811:25–30.
Delgado Alves J, Ames PR, Donohue S et al. Antibodies to highdensity lipoprotein and beta2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum 2002;46:2686–2694.
Batuca JR, Ames PR, Isenberg DA et al. Antibodies toward high-density lipoprotein components inhibit paraoxonase activity in patients with systemic lupus erythematosus. Ann NY Acad Sci 2007;1108:137–146.
Ames PR, Matsuura E, Batuca JR et al. High-density lipoprotein inversely relates to its specific autoantibody favoring oxidation in thrombotic primary antiphospholipid syndrome. Lupus 2010;19:711–716.
Srivastava R, Yu S, Parks BW et al. Autoimmune-mediated reduction of high-density lipoprotein-cholesterol and paraoxonase 1 activity in systemic lupus erythematosus-prone gld mice. Arthritis Rheum 2011;63:201–211.
Montecucco F, Vuilleumier N, Pagano S et al. Anti-Apolipoprotein A-1 auto-antibodies are active mediators of atherosclerotic plaque vulnerability. Eur Heart J 2011;32:412–421.
Pagano S, Satta N, Werling D et al. Anti-apolipoprotein A-1 IgG in patients with myocardial infarction promotes inflammation through TLR2/CD14 complex. J Intern Med 2012;272:344–357.
Montecucco F, Braunersreuther V, Burger F et al. Anti-apoA-1 auto-antibodies increase mouse atherosclerotic plaque vulnerability, myocardial necrosis and mortality triggering TLR2 and TLR4. Thromb Haemost 2015;114:410–422.
Vuilleumier N, Rossier MF, Pagano S et al. Anti-apolipoprotein A-1 IgG as an independent cardiovascular prognostic marker affecting basal heart rate in myocardial infarction. Eur Heart J 2010;31:815–823.
Zhang GQ, Zhang W . Heart rate, lifespan, and mortality risk. Ageing Res Rev 2009;8:52–60.
Menown IB, Davies S, Gupta S et al. Resting heart rate and outcomes in patients with cardiovascular disease: where do we currently stand? Cardiovasc Ther 2013;31:215–223.
Rossier MF, Pagano S, Python M et al. Antiapolipoprotein A-1 IgG chronotropic effects require nongenomic action of aldosterone on L-type calcium channels. Endocrinology 2012;153:1269–1278.
Mannic T, Satta N, Pagano S et al. CD14 as a mediator of the mineralocorticoid receptor-dependent anti-apolipoprotein A-1 IgG chronotropic effect on cardiomyocytes. Endocrinology 2015;156:4707–4719.
Mackness B, Hunt R, Durrington PN et al. Increased immunolocalization of paraoxonase, clusterin, and apolipoprotein A-I in the human artery wall with the progression of atherosclerosis. Arterioscler Thromb Vasc Biol 1997;17:1233–1238.
Wick PA, Mombelli A, Pagano S et al. Anti-apolipoprotein A-1 autoantibodies as biomarker for atherosclerosis burden in patients with periodontitis. J Periodontal Res 2013;48:350–356.
Pruijm M, Schmidtko J, Aho A et al. High prevalence of anti-apolipoprotein/A-1 autoantibodies in maintenance hemodialysis and association with dialysis vintage. Ther Apher Dial 2012;16:588–594.
El-Lebedy D, Rasheed E, Kafoury M et al. Anti-apolipoprotein A-1 autoantibodies as risk biomarker for cardiovascular diseases in type 2 diabetes mellitus. J Diabetes Complications 2016;30:580–585.
Rodríguez-Carrio J, Alperi-López M, López P et al. Antibodies to high-density lipoproteins are associated with inflammation and cardiovascular disease in rheumatoid arthritis patients. Transl Res 2015;166:529–539.
Rodríguez-Carrio J, López-Mejías R, Alperi-López M et al. PON activity is modulated by rs662 polymorphism and IgG anti-HDL antibodies in rheumatoid arthritis patients: potential implications for CV disease. Arthritis Rheumato, ; e-pub ahead of print 27 January 2016; doi:10.1002/art.39609.
Vuilleumier N, Charbonney E, Fontao L et al. Anti-(apolipoprotein A-1) IgGs are associated with high levels of oxidized low-density lipoprotein in acute coronary syndrome. Clin Sci (Lond) 2008;115:25–33.
Keller PF, Pagano S, Roux-Lombard P et al. Autoantibodies against apolipoprotein A-1 and phosphorylcholine for diagnosis of non-ST-segment elevation myocardial infarction. J Intern Med 2012;271:451–462.
Vuilleumier N, Montecucco F, Spinella G et al. Serum levels of anti-apolipoprotein A-1 auto-antibodies and myeloperoxidase as predictors of major adverse cardiovascular events after carotid endarterectomy. Thromb Haemost 2013;109:706–715.
Quercioli A, Montecucco F, Galan K et al. Anti-apolipoprotein A-1 IgG levels predict coronary artery calcification in obese but otherwise healthy individuals. Mediators Inflamm 2012;2012:243158.
Vuilleumier N, Bas S, Pagano S et al. Anti-apolipoprotein A-1 IgG predicts major cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum 2010;62:2640–2650.
Finckh A, Courvoisier DS, Pagano S et al. Anti-apolipoprotein A-1 IgG predicts major cardiovascular events in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:817–825.
Rubini Gimenez M, Pagano S, Virzi J et al. Diagnostic and prognostic value of autoantibodies anti-apolipoprotein A-1 and anti-phosphorylcholine in acute non-ST elevation myocardial infarction. Eur J Clin Invest 2015;45:369–379.
Vuilleumier N, Pagano S, Lahlou K et al. Head-to-head comparison of auto-antibodies for cardiovascular outcome prediction after myocardial infarction: a prospective study. J Clinic Experiment Cardiol 2011;2:169.
Vuilleumier N, Montecucco F, Hartley O . Autoantibodies to apolipoprotein A-1 as a biomarker of cardiovascular autoimmunity. World J Cardiol 2014;6:314–326.
Satta N, Vuilleumier N . Auto-antibodies as possible markers and mediators of ischemic, dilated, and rhythmic cardiopathies. Curr Drug Targets 2015;16:342–360.
Acknowledgements
This work was supported by the Ministry of Education and Sciences, Russian Federation (Project# RFMEFI61614X0010).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Chistiakov, D., Orekhov, A. & Bobryshev, Y. ApoA1 and ApoA1-specific self-antibodies in cardiovascular disease. Lab Invest 96, 708–718 (2016). https://doi.org/10.1038/labinvest.2016.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.56
This article is cited by
-
Higher chocolate intake is associated with longer telomere length among adolescents
Pediatric Research (2020)
-
Antibodies towards high-density lipoprotein components in patients with psoriasis
Archives of Dermatological Research (2020)
-
Modifications of human plasma apolipoprotein A1 in systemic autoimmune diseases and myocardial infarction: a comparative study
Journal of Proteins and Proteomics (2019)
