
Abstract
Chronic obstructive pulmonary disease (COPD) is a disease common in elderly people, characterized by progressive destruction of lung parenchyma and chronic inflammation of the airways. The pathogenesis of COPD remains unclear, but recent studies suggest that oxidative stress-induced apoptosis in alveolar cells contributes to emphysematous lung destruction. The proteasome is a multicatalytic enzyme complex that plays a critical role in proteostasis by rapidly destroying misfolded and modified proteins generated by oxidative and other stresses. Proteasome activity decreases with aging in many organs including lungs, and an age-related decline in proteasomal function has been implicated in various age-related pathologies. However, the role of the proteasome system in the pathogenesis of COPD has not been investigated. Recently, we have established a transgenic (Tg) mouse model with decreased proteasomal chymotrypsin-like activity, showing age-related phenotypes. Using this model, we demonstrate here that decreased proteasomal function accelerates cigarette smoke (CS)-induced pulmonary emphysema. CS-exposed Tg mice showed remarkable airspace enlargement and increased foci of inflammation compared with wild-type controls. Importantly, apoptotic cells were found in the alveolar walls of the affected lungs. Impaired proteasomal activity also enhanced apoptosis in cigarette smoke extract (CSE)-exposed fibroblastic cells derived from mice and humans in vitro. Notably, aggresome formation and prominent nuclear translocation of apoptosis-inducing factor were observed in CSE-exposed fibroblastic cells isolated from Tg mice. Collective evidence suggests that CS exposure and impaired proteasomal activity coordinately enhance apoptotic cell death in the alveolar walls that may be involved in the development and progression of emphysema in susceptible individuals such as the elderly.
Similar content being viewed by others

Main
Chronic obstructive pulmonary disease (COPD) involves progressive airflow obstruction and airway inflammation, and represents one of the leading causes of morbidity and mortality throughout the world.1, 2 The primary risk factor for COPD is chronic cigarette smoke (CS) exposure; other risk factors for COPD include wood fuel smoke, environmental air pollutants, respiratory infection, and aging.3 The pathogenesis of COPD remains poorly understood, but involves persistent inflammation, oxidative stress, impaired cell repair and cell death, and destruction of extracellular matrix.4, 5 In addition, recent studies have suggested additional mechanisms involving altered protein homeostasis (proteostasis),6, 7 such as endoplasmic reticulum (ER) stress, inhibition of the ubiquitin–proteasome system, and autophagy.8, 9, 10, 11, 12
The proteasome is a multicatalytic enzyme complex responsible for the maintenance of cellular homeostasis and plays an essential role in numerous biologic processes such as cell proliferation, cell cycling, gene transcription, apoptosis, antioxidant responses, and immune reactions.13 In addition, the proteasome serves as a protein quality control system by rapidly destroying misfolded and oxidatively modified proteins.14 Therefore, impaired proteasome function can result in the accumulation of ubiquitinated and altered proteins, resulting in cellular dysfunction and cell death.15 Physiologically, proteasomal activity is decreased with age,16, 17 and an age-associated decline in proteasome function is widely assumed to contribute to the development of age-related pathology and to the aging process itself.17 In COPD patients, an age-associated decline in proteasomal activity and subunit expression has been found to inversely correlate with lung function.9 However, the role of the proteasome in the pathogenesis of COPD has not been experimentally addressed.
Recently, we have established a transgenic (Tg) mouse model with decreased proteasomal chymotrypsin-like activity.18 Tg mice exhibited a shortened lifespan and age-related phenotypes with the accumulation of polyubiquitinated and oxidized proteins. Tg mice <12 weeks of age displayed no apparent histological abnormality. However, after 24 weeks of age, they exhibited lordokyphosis (curvature of the spinal column) and a loss of subcutaneous adipose tissue and skeletal muscle mass, both prominent features of normal aging. Tg mice died without any gross pathological changes including emphysema, cancer, and cardiovascular diseases. Interestingly, Tg mice were susceptible to obesity induced by high-fat diet and hepatic steatosis, similar to metabolic abnormalities typically seen in aged mice.18 Using this model, we provide in vivo evidence that decreased proteasomal function accelerates CS-induced pulmonary emphysema. Moreover, we provide in vitro evidence that impaired proteasomal activity promotes cigarette smoke extract (CSE)-induced aggresome formation and increases apoptotic cell death mediated by apoptosis-inducing factor (AIF).
MATERIALS AND METHODS
Mice and CS Exposure Experiments
Tg mice with decreased proteasomal chymotrypsin-like activity were established as previously described.18 C57BL/6 mice were used as wild-type (WT) controls. Tg and WT mice were housed on a 12-h light/dark cycle in climate-controlled, pathogen-free barrier facilities. Using the SIS-CS system (Shibata Scientific Technology, Tokyo, Japan), 9-week-old Tg and WT mice were exposed to mainstream CS (5% CS, 60 min/day for 10 consecutive days or 5 days/week for 12 weeks), as previously reported.19 CS was generated from commercially available filtered cigarettes (Marlboro, 12 mg tar/1.0 mg nicotine; Philip Morris, Richmond, VA, USA). Age-matched, nontreated control mice were exposed to room air. All animal experiments were done according to the Guidelines for the Care and Use of Laboratory Animals at Hokkaido University Graduate School of Medicine.
Cell Isolation and Cell Culture
Primary lung fibroblastic cells (LFCs) and alveolar epithelial cells (AECs) from C57BL/6 mice were purchased from Cell Biologics (Chicago, IL, USA). Normal human lung fibroblastic cells (NHLFs) were purchased from Lonza Japan (Tokyo, Japan). Primary mouse fibroblastic cells (FCs) were isolated from abdominal subcutaneous tissues of Tg or WT mice using collagenase digestion. LFCs, AECs, and NHLFs were grown in media recommended by the manufacturers, and FCs from Tg or WT mice were grown in DMEM supplemented with 20% fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin in an atmosphere of 5% CO2 and 95% O2 at 37 °C, and assayed during passages 3 to 4. An equivalent number of cells (4.0 × 104/well in a 12 multiwell plate) suspended in DMEM medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin were treated with CSE (1.0 to 3.0%) for 16 h or PS-341 (10 to 100 nM) for 48 h. The concentration of CSE or PS-341 was adjusted for each fibroblast strain to yield ∼10–30% annexin V-positive cells after a single administration. For treatment with both reagents, cells were incubated with PS-341 for 32 h, and then CSE was added to the plates for 16 h. For nontreated controls, we used PBS and DMEM medium for CSE and PS-341, respectively.
Histological Analysis
The lungs were fixed with buffered 10% formalin via endotracheal instillation at a pressure of 25 cmH2O. After paraffin embedding, 4 μm sections were stained with hematoxylin and eosin (H&E). Alveolar size of the lung was assessed by the determination of the mean linear intercepts (Lm). Lm was calculated based on 20 randomly selected fields in each section (in total 50 fields/mouse) at × 200 magnification with two crossed test lines.19 The intercepts of alveolar walls with these lines were counted. Fields with airways or blood vessels were not used for morphometry. To evaluate the extent of inflammation in the lungs, the inflammatory foci in which >50 lymphocytes formed an aggregate were counted. TdT-mediated dUTP nick-end labeling (TUNEL) staining was performed using the In situ Apoptosis Detection Kit (Takara, Shiga, Japan) according to the manufacturer’s protocol. To detect aggresomes, FCs were cultured in chamber slides, and the slides were stained according to the manufacturer’s instructions (Proteostat Aggresome Detection Kit, Enzo Life Sciences, Lausen, Switzerland). For immunofluorescence staining for AIF, FCs on chamber slides were washed twice in PBS, fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.1% Triton-X in PBS for 5 min. After washing, the slides were incubated in PBS with 10% goat serum for 1 h at room temperature, followed by overnight incubation with anti-AIF antibody (Ab) (Cell Signaling Technology, Tokyo, Japan) at 4 °C. Sections were labeled with Alexa 488-conjugated anti-rabbit IgG (Invitrogen) and mounted with DAPI-containing mounting medium (Vector Laboratories, Burlingame, CA, USA) for nuclear staining. Nuclear translocation of AIF was quantitated by counting the percentage of cells with nuclear AIF in 10 randomly selected fields.
Preparation of CSE
CS exposure was modeled in vitro by preparing CSE according to the published method.20, 21 Briefly, one cigarette per trial was fixed horizontally to be burned, and the main stream of the smoke was aspirated at a flow rate of 1.050 l/min that was strictly regulated by the KOFLOC mass flow controller (MODEL 8300 series, Kojima Instruments, Kyoto, Japan). The main stream of the smoke was passed through a Cambridge glass fiber filter to remove the tar phase and nicotine from the CS, and the remaining gas phase of the smoke was directly collected in a cooled vessel at −80 °C with dry ice–acetone and stored at −80 °C.
Flow Cytometry
For the detection of aggresomes, cells were stained according to the manufacturer’s instructions (Proteostat Aggresome Detection Kit). Data were evaluated by relative values, setting the mean fluorescence intensity (MFI) of WT controls as 1. Surface exposure of phosphatidylserine in apoptotic cells was quantitatively detected using the annexin V-FITC Apoptosis Kit (BioVision, Mountain View, CA, USA). Flow cytometric analysis was performed using FACSCalibur and CellQuest software (BD Bioscience, Tokyo, Japan).
Western Blot Analysis
FCs were lysed in a buffer containing 150 mM NaCl, 20 mM Tris-HCl (pH 7.5), 0.2% NP-40, and 1 mM DTT, and centrifuged at 15 000 g for 10 min. The supernatants (10 μg aliquot of total protein) were subjected to SDS-PAGE and blotted to nitrocellulose membranes. The blots were probed with Abs, and reacted with horseradish peroxidase-conjugated anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA) for immunodetection. The immune complexes were visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ, USA) and analyzed by Image Gauge software (Fujifilm, Tokyo, Japan). Abs for GRP78 and actin were purchased from Abcam Japan (Tokyo, Japan). Abs for caspase-3 and AIF were purchased from Cell Signaling Technology.
Statistical Analysis
We performed statistical analyses for two unmatched groups with the unpaired two-tail Student’s t-test. For the analysis of three or more unmatched groups, one-way ANOVA with multiple comparisons/post hoc tests was performed. P-values of <0.05 were considered significant.
RESULTS
Decreased Proteasomal Function Accelerates CS-Induced Pulmonary Emphysema In Vivo
To investigate the involvement of proteasomal function in the pathogenesis of COPD, Tg mice with decreased proteasomal chymotrypsin-like activity were exposed to CS. Although a slight increase in the airspace was observed in the lung tissue of CS-exposed WT mice, the airspace was remarkably enlarged in CS-exposed Tg mice. Lm was significantly increased in CS-exposed Tg mice (Figures 1a and b). In addition, the number of foci with inflammatory cell infiltration was increased in CS-exposed Tg mice (Figures 1a and c). As recent studies from both animal models and COPD patients suggest that apoptosis may represent an important factor in the pathogenesis of COPD,22, 23, 24 we next performed TUNEL staining to detect apoptotic cells in the lungs of CS-exposed mice. As shown in Figures 1d and e, apoptotic cells were abundantly found in the affected lungs of CS-exposed Tg mice. These results suggest that decreased proteasomal function accelerates CS-induced apoptotic cell death in the lungs that then leads to morphologic features characteristic of pulmonary emphysema such as airspace enlargement and destruction of alveolar walls.

CS-induced pulmonary emphysema in mice with decreased proteasomal function. (a) Representative photographs of lung tissues in WT and Tg mice exposed or not exposed to CS. (b) Mean linear intercept (Lm) of the lung. (c) Number of inflammatory foci in the lung. Foci with the infiltration of >50 lymphocytes were counted. HPF, high-power field. (d) Apoptotic cells in the lung of CS-exposed mice. Apoptotic cells (arrows) were identified with TUNEL staining. (e) Number of apoptotic cells in the lung of CS-exposed mice. TUNEL-positive cells were counted in square 200 μm. All data are presented as means±s.d. (n=10). *P<0.05; **P<0.01.
CSE-Induced Apoptotic Cell Death Is Increased in Fibroblastic Cells with Impaired Proteasomal Function
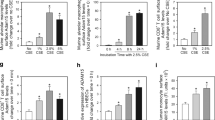
Fibroblasts are the main cell type in the lung interstitium, and play a pivotal role in the repair, remodeling, and maintenance of alveolar structure.25 To examine whether decreased proteasomal activity aggravates CSE-induced apoptosis, we performed in vitro experiments using primary cells derived from mouse lungs. First, we examined the induction of apoptosis by CSE and/or PS-341 in LFCs and AECs. As shown in Figure 2a, PS-341 treatment increased annexin V-positive cells to a similar extent in LFCs and AECs. However, CSE treatment did not induce cell death in AECs in the concentrations tested.

CSE-induced apoptotic cell death in fibroblastic cells with impaired proteasomal function. (a) Sensitivity to CSE and PS-341 in LFCs and AECs. (b–d) Apoptotic cell death in CSE- and/or PS-341-exposed LFCs (b), AECs (c), NHLFs (d), and primary mouse FCs derived from subcutaneous tissues (e). Cells were incubated in culture medium containing indicated concentrations of PS-341 and/or CSE. (f) Apoptotic cell death in CSE-exposed FCs from WT and Tg mice. FCs from WT and Tg mice were exposed to 1% CSE for 16 h. All data are presented as means±s.d. from three independent experiments (n=5 per group). *P<0.05; **P<0.01; ***P<0.001.
Next, LFCs and AECs were incubated in CSE-containing medium (1.0%) for 16 h with or without prior treatment with proteasome inhibitor PS-341 (50 nM) for 32 h. When LFCs were treated with both CSE and PS-341, the number of annexin V-positive cells was significantly increased in comparison with control and cells treated with CSE or PS-341 alone (Figure 2b). The proportion of propidium iodide (PI)-positive cells in annexin V-positive cells was also significantly increased, indicating that either late apoptotic or necrotic cells were induced in CSE-exposed LFCs with decreased proteasomal function. In contrast, consistent with the observation that CSE failed to induce apoptosis in AECs, the number of annexin V-positive cells and PI-positive cells was almost the same in cells treated with PS-341 alone and those treated with both PS-341 and CSE (Figure 2c). When we used human NHLFs, apoptotic cell death was significantly increased in cells treated with both CSE and PS-341, in comparison with control and cells treated with CSE or PS-341 alone (Figure 2d). Similar results were obtained with mouse FCs derived from subcutaneous tissues (Figure 2e), suggesting that fibroblasts are sensitive to CSE- and PS-341-induced apoptosis in both humans and mice, and presumably regardless of their tissue origin. It appears that CS exposure and impaired proteasomal activity coordinately induce apoptotic cell death in fibroblasts.
Finally, we exposed FCs from WT or Tg mice to 1% CSE for 16 h. As shown in Figure 2f, the number of annexin V-positive cells was significantly increased in FCs from both WT and Tg mice compared with non-CSE-exposed FCs. Interestingly, annexin V-positive cells were more abundant in Tg mice-derived FCs than in WT mice-derived FCs (Figure 2f, upper panel). The proportion of PI-positive cells in annexin V-positive cells was also significantly increased in CSE-exposed FCs from Tg mice (Figure 2f, lower panel). Taken together, these results indicate that the impairment of proteasomal function potentiates CSE-induced cell death in FCs, regardless of whether it is induced by genetic manipulation or by a specific inhibitor.
CSE-Induced Aggresome Formation Is Enhanced in FCs with Impaired Proteasomal Function
Each CS contains >1014 free radicals, including reactive oxygen species (ROS), that impose strong oxidative stress resulting in the formation of misfolded and aggregated proteins.26, 27 Misfolded or abnormal proteins are either refolded by chaperones or degraded by proteasomes. However, when proteasomal function is impaired, misfolded proteins form aggresomes, resulting in cellular toxicity.28, 29 Consistent with these observations, we have previously demonstrated that FCs from Tg mice with impaired proteasomal function show enhanced aggresome formation.18
We therefore examined the effect of CSE exposure on aggresome formation in FCs from WT and Tg mice (Figure 3). When FCs were incubated in CSE-containing medium, aggresome formation occurred in FCs from both WT and Tg mice. However, aggresome formation was much more prominent in FCs from Tg mice, indicating that decreased proteasomal activity enhances aggresome formation induced by oxidative stress.

CSE-induced aggresomes in FCs with impaired proteasomal function. Aggresome formation was detected by immunohistochemistry (a) and flow cytometry (b). FCs from WT and Tg mice were cultured in CSE-containing medium (1.0%) and subjected to the experiments. (a) Representative photographs. Red, aggresomes; blue, nuclear DAPI stain. (b) Flow cytometry data were evaluated by relative values, setting the MFI of WT controls as 1. Data were from three independent experiments. *P<0.05.
Expression of ER Stress and Apoptosis-Related Molecules in CSE-Exposed FCs with Impaired Proteasomal Function
Oxidative stress causes the accumulation of misfolded and unfolded proteins in the ER.30 The resultant ER stress induces expression of ER chaperones, inhibits translation, and promotes the retrograde transport of misfolded proteins to the cytosol for degradation by the proteasome (a process known as ER-associated degradation (ERAD)),31 thus restoring protein homeostasis. Impaired proteasomal activity disrupts ERAD, resulting in increased ER stress, and when stress persists and becomes more severe, it can trigger apoptosis.15, 32 In addition, oxidative stress can induce the disintegration of mitochondrial membrane, leading to mitochondria-associated events such as apoptosis.33
To examine the apoptosis pathways involved in CSE-induced cell death prominently observed in FCs with decreased proteasomal function, we analyzed the expression of the following molecules involved in ER stress and apoptosis: glucose-regulated protein 78 (GRP78), a major ER-resident chaperone; caspase-3 and cleaved caspase-3, key executioners of apoptosis wherein mitochondrial and ER stress pathways converge; and AIF.34, 35, 36 Expression of GRP78 was upregulated in CSE-exposed FCs from Tg mice (Figure 4a). However, cleaved caspase-3 showed no significant increase in CSE-exposed FCs from WT or Tg mice (Figure 4a), indicating that apoptosis was mainly induced by a caspase-independent mechanism. Notably, expression of AIF was increased, and prominent nuclear translocation of AIF was frequently observed in CSE-exposed FCs from Tg mice compared with those from WT mice (Figure 4b).


Expression of ER stress and apoptosis-related molecules in CSE-exposed FCs with impaired proteasomal function. (a) Western blot analysis of GRP78, caspase-3, cleaved caspase-3, and AIF in CSE-exposed and -nonexposed FCs. Analysis was performed for FCs from WT and Tg mice. (b) Nuclear translocation of AIF was detected by immunofluorescence staining. Dense accumulation of AIF was observed in CSE-exposed FCs from Tg mice (arrow). Nuclear translocation of AIF was quantitated by counting the percentage of cells with nuclear AIF in 10 randomly selected fields. *P<0.05; **P<0.01.
DISCUSSION
Recent studies from both animal models and patients suggest that oxidative stress-induced apoptosis is an important factor in the pathogenesis of COPD and contributes to emphysematous lung destruction in response to CS.22, 23, 24 On the other hand, a significant increase in the prevalence of COPD among the elderly is assumed to be attributable to age-associated changes in the lung.37 Decreased proteasomal activity with aging has been widely documented in many organs including lungs.16, 38, 39 An age-related decrease in proteasome activity weakens cellular capacity to remove damaged proteins and favors the development of diseases.38 In this study, taking advantage of the mouse model developed in our laboratory,18 we provided direct in vivo evidence for the first time that decreased proteasomal function accelerates CS-induced pulmonary emphysema (Figure 1). Moreover, FCs with impaired proteasomal activity showed increased susceptibility to CS-induced apoptotic cell death in vitro (Figures 2 and 4). These observations indicate strongly that an age-related decrease in proteasomal function contributes to CS-induced alveolar wall damages and to the development of COPD.
Oxidative stress elicits ER stress that triggers the unfolded protein response (UPR) to restore protein homeostasis. GRP78 acts as a master regulator in this response to protect cells against the toxic buildup of misfolded proteins.35, 40 Misfolded proteins are then degraded by proteasomes via ERAD.31, 41 Thus, impaired proteasomal activity disrupts UPR, causing aggregation and accumulation of misfolded proteins, G2/M cell cycle arrest, and eventually cell death.15, 28 It has been reported that CS causes ER stress in the lungs of patients with COPD, implicating ER stress in its pathogenesis.42, 43 CSE-exposed FCs from Tg mice showed increased expression of GRP78 and aggresome formation (Figures 3 and 4a). These results suggest that CS exposure and decreased proteasomal function additively or possibly even synergistically cause strong ER stress in the lung, overwhelming the capacity of the proteostasis system and ultimately leading to cell death and tissue destruction.
Along with the ER, mitochondria are prime targets of oxidative stress; they play an important role in both caspase-dependent and -independent apoptotic pathways.36, 44 A major caspase-independent mechanism of cell death is regulated by AIF, a flavoprotein normally localized in the outer mitochondrial membrane.36 Upon release from the mitochondria, AIF translocates to the nucleus where it induces DNA fragmentation and chromatin condensation.45 In the present study, there was no significant activation of caspase-3 in CSE-exposed FCs (Figure 4a). Instead, we found increased expression of AIF and its nuclear translocation in CSE-exposed FCs from Tg mice (Figure 4b). This is consistent with the previous observation that proteasomal inhibition reduces AIF ubiquitination,46 alters mitochondrial membrane potentials, and induces the nuclear translocation of AIF through downregulation of ERK and Akt/mTOR pathways.47
Increased alveolar cell apoptosis is observed in the emphysematous lung.48, 49, 50, 51 The loss of alveolar wall structure associated with emphysema may be precipitated by an imbalance of cell proliferation and death, with excessive apoptosis leading to emphysematous lesions.52 Many cells within the alveolar wall are sensitive to CS, including immune cells, endothelial and epithelial cells, and fibroblasts.53, 54, 55, 56, 57 Both epithelial cells and fibroblasts rapidly undergo apoptosis after CS exposure.56, 57, 58 Thus, the coordinated loss of structural and repair cells in alveoli coupled with chronic inflammation presumably constitutes the central events during the development and progression of emphysema.23, 59 Interestingly, CSE-induced apoptosis was observed in LFCs, but not in AECs, in this study (Figure 2). The observation that AECs were not sensitive to CSE-induced apoptosis may appear inconsistent with previous studies. However, experimental designs including the source of cells, CSE concentrations, and incubation time vary greatly among individual reports.58, 60, 61, 62 To our knowledge, few studies have addressed a cell type-specific difference in sensitivity to CSE-induced apoptosis. In this study, we compared the sensitivity to CSE and PS-341 using mouse primary cells with the same C57BL/6-background, revealing that LFCs exhibit higher sensitivity to CSE than AECs in the concentrations tested. Fibroblasts are the main cell type in the lung interstitium and provide structural support to the alveolar compartment; they are believed to be an important target of CS-induced damage.57, 63, 64, 65
Failure to maintain protein homeostasis causes many human diseases.66 In addition, aging is accompanied by a gradual, yet progressive, decrease in proteasomal function. Our findings provide important insights into the pathogenesis of age-related emphysema, and should contribute to the development of a new approach to the prevention and treatment of COPD.
References
Buist AS, McBurnie MA, Vollmer WM et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet 2007;370:741–750.
Decramer M, Janssens W, Miravitlles M . Chronic obstructive pulmonary disease. Lancet 2012;379:1341–1351.
Mannino DM, Buist AS . Global burden of COPD: risk factors, prevalence, and future trends. Lancet 2007;370:765–773.
Barnes PJ, Shapiro SD, Pauwels RA . Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003;22:672–688.
Yoshida T, Tuder RM . Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007;87:1047–1082.
Bodas M, Tran I, Vij N . Therapeutic strategies to correct proteostasis-imbalance in chronic obstructive lung diseases. Curr Mol Med 2012;12:807–814.
Min T, Bodas M, Mazur S et al. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J Mol Med (Berl) 2011;89:577–593.
van Rijt SH, Keller IE, John G et al. Acute cigarette smoke exposure impairs proteasome function in the lung. Am J Physiol Lung Cell Mol Physiol 2012;303:L814–L823.
Malhotra D, Thimmulappa R, Vij N et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2-regulated proteasomal activity. Am J Respir Crit Care Med 2009;180:1196–1207.
Somborac-Bacura A, van der Toorn M, Franciosi L et al. Cigarette smoke induces endoplasmic reticulum stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp Physiol 2013;98:316–325.
Chen ZH, Kim HP, Sciurba FC et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008;3:e3316.
Chen ZH, Lam HC, Jin Y et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA 2010;107:18880–18885.
Coux O, Tanaka K, Goldberg AL . Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 1996;65:801–847.
Poppek D, Grune T . Proteasomal defense of oxidative protein modifications. Antioxid Redox Signal 2006;8:173–184.
Chauhan D, Singh A, Brahmandam M et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 2008;111:1654–1664.
Chondrogianni N, Gonos ES . Proteasome dysfunction in mammalian aging: steps and factors involved. Exp Gerontol 2005;40:931–938.
Dahlmann B . Role of proteasomes in disease. BMC Biochem 2007;8:S3.
Tomaru U, Takahashi S, Ishizu A et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol 2012;180:963–972.
Suzuki M, Betsuyaku T, Ito Y et al. Curcumin attenuates elastase- and cigarette smoke-induced pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol 2009;296:L614–L623.
Asano H, Horinouchi T, Mai Y et al. Nicotine- and tar-free cigarette smoke induces cell damage through reactive oxygen species newly generated by PKC-dependent activation of NADPH oxidase. J Pharmacol Sci 2012;118:275–287.
Noya Y, Seki K, Asano H et al. Identification of stable cytotoxic factors in the gas phase extract of cigarette smoke and pharmacological characterization of their cytotoxicity. Toxicology 2013;314:1–10.
Sakao S, Tatsumi K, Hashimoto T et al. Vascular endothelial growth factor and the risk of smoking-related COPD. Chest 2003;124:323–327.
Demedts IK, Demoor T, Bracke KR et al. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 2006;7:53.
Park JW, Ryter SW, Kyung SY et al. The phosphodiesterase 4 inhibitor rolipram protects against cigarette smoke extract-induced apoptosis in human lung fibroblasts. Eur J Pharmacol 2013;706:76–83.
Tuder RM, Yoshida T, Fijalkowka I et al. Role of lung maintenance program in the heterogeneity of lung destruction in emphysema. Proc Am Thorac Soc 2006;3:673–679.
Yoshiyama S, Horinouchi T, Miwa S et al. Effect of cigarette smoke components on vascular smooth muscle cell migration toward platelet-derived growth factor BB. J Pharmacol Sci 2011;115:532–535.
Kawaguchi Y, Kovacs JJ, McLaurin A et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003;115:727–738.
Bence NF, Sampat RM, Kopito RR . Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001;292:1552–1555.
Olzmann JA, Li L, Chin LS . Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem 2008;15:47–60.
Lin JH, Walter P, Yen TS . Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 2008;3:399–425.
Kincaid MM, Cooper AA . ERADicate ER stress or die trying. Antioxid Redox Signal 2007;9:2373–2387.
McCullough KD, Martindale JL, Klotz LO et al. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001;21:1249–1259.
Simon HU, Haj-Yehia A, Levi-Schaffer F . Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000;5:415–418.
Salvesen GS, Dixit VM . Caspases: intracellular signaling by proteolysis. Cell 1997;91:443–446.
Rao RV, Bredesen DE . Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol 2004;16:653–662.
Susin SA, Lorenzo HK, Zamzami N et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999;397:441–446.
Lowery EM, Brubaker AL, Kuhlmann E et al. The aging lung. Clin Interv Aging 2013;8:1489–1496.
Low P . The role of ubiquitin-proteasome system in ageing. Gen Comp Endocrinol 2011;172:39–43.
Breusing N, Arndt J, Voss P et al. Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mech Ageing Dev 2009;130:748–753.
Wang M, Wey S, Zhang Y et al. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal 2009;11:2307–2316.
Vembar SS, Brodsky JL . One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 2008;9:944–957.
Kelsen SG, Duan X, Ji R et al. Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol 2008;38:541–550.
Adair-Kirk TL, Atkinson JJ, Senior RM . Smoke particulates stress lung cells. Nat Med 2008;14:1024–1025.
Gross A, McDonnell JM, Korsmeyer SJ . BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999;13:1899–1911.
Joza N, Susin SA, Daugas E et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001;410:549–554.
Zaman F, Menendez-Benito V, Eriksson E et al. Proteasome inhibition up-regulates p53 and apoptosis-inducing factor in chondrocytes causing severe growth retardation in mice. Cancer Res 2007;67:10078–10086.
Ko JK, Choi CH, Kim YK et al. The proteasome inhibitor MG-132 induces AIF nuclear translocation through down-regulation of ERK and Akt/mTOR pathway. Neurochem Res 2011;36:722–731.
Segura-Valdez L, Pardo A, Gaxiola M et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000;117:684–694.
Kasahara Y, Tuder RM, Cool CD et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001;163:737–744.
Imai K, Mercer BA, Schulman LL et al. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J 2005;25:250–258.
Hodge S, Hodge G, Holmes M et al. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J 2005;25:447–454.
Calabrese F, Giacometti C, Beghe B et al. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res 2005;6:14.
Zavitz CC, Gaschler GJ, Robbins CS et al. Impact of cigarette smoke on T and B cell responsiveness. Cell Immunol 2008;253:38–44.
Damico R, Simms T, Kim BS et al. p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: inhibitory effects of macrophage migration inhibitor factor. Am J Respir Cell Mol Biol 2011;44:323–332.
Pace E, Ferraro M, Siena L et al. Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 2008;124:401–411.
Baglole CJ, Bushinsky SM, Garcia TM et al. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol 2006;291:L19–L29.
Carnevali S, Petruzzelli S, Longoni B et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2003;284:L955–L963.
Tagawa Y, Hiramatsu N, Kasai A et al. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic Biol Med 2008;45:50–59.
Park JW, Ryter SW, Choi AM . Functional significance of apoptosis in chronic obstructive pulmonary disease. COPD 2007;4:347–353.
An CH, Wang XM, Lam HC et al. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol 2012;303:L748–L757.
Yuan T, Luo BL, Wei TH et al. Salubrinal protects against cigarette smoke extract-induced HBEpC apoptosis likely via regulating the activity of PERK-eIF2alpha signaling pathway. Arch Med Res 2012;43:522–529.
Bazzini C, Rossetti V, Civello DA et al. Short- and long- term effects of cigarette smoke exposure on glutathione homeostasis in human bronchial epithelial cells. Cell Physiol Biochem 2013;32:129–145.
Chen LJ, Zhao Y, Gao S et al. Downregulation of lysyl oxidase and upregulation of cellular thiols in rat fetal lung fibroblasts treated with cigarette smoke condensate. Toxicol Sci 2005;83:372–379.
Holz O, Zuhlke I, Jaksztat E et al. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J 2004;24:575–579.
Ishii T, Matsuse T, Igarashi H et al. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol 2001;280:L1189–L1195.
Buchberger A, Bukau B, Sommer T . Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 2010;40:238–252.
Acknowledgements
We are grateful to Ms Junko Tamba, Department of Pathology, Hokkaido University Graduate School of Medicine, for technical support. We also thank all the staff of the Institute for Animal Experimentation, Hokkaido University Graduate School of Medicine, for maintenance of mice. This work was supported in part by Takeda Science Foundation and Grants-in-Aid for Scientific Research from The Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This paper reveals that the pathogenesis of chronic obstructive pulmonary disease (COPD) is related to oxidative stress-induced apoptosis in alveolar cells. Transgenic (Tg) mice with decreased proteasomal chymotrypsin-like activity experience accelerated development of cigarette smoke (CS)-induced pulmonary emphysema. Morphologically, CS-exposed Tg mice show remarkable air space enlargement, increased foci of inflammation and apoptotic cells in the alveolar walls of the affected lungs.
Rights and permissions
About this article

Cite this article
Yamada, Y., Tomaru, U., Ishizu, A. et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice. Lab Invest 95, 625–634 (2015). https://doi.org/10.1038/labinvest.2015.43
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2015.43
This article is cited by
-
Augmenting autophagy for prognosis based intervention of COPD-pathophysiology
Respiratory Research (2017)
