
Abstract
The molecular impact of diabetes mellitus on prostate gland has not been elucidated. In this study, we performed a whole-genome cDNA microarray analysis using a streptozotocin-induced diabetic rat model to identify the effects of diabetes on the gene expression profiles in prostate. Our study shows that diabetes causes changes in the expression of multiple genes, particularly those related to cell proliferation and differentiation, oxidative stress, DNA damage repair, cell cycle checkpoints, angiogenesis and apoptosis. These findings were confirmed by real-time polymerase chain reaction and immunohistochemical staining using rat and human prostate tissue. We also used a cell culture model (human normal prostatic RWPE-1 cell line) to study the direct effect of high glucose. We found that high glucose caused increased intracellular oxidative stress and DNA damage, as well as downregulation of anti-oxidative enzymes and DNA damage repair genes MRE11 and XRCC3. Our findings provide important insights into understanding the pathogenesis of the diabetes-induced changes in prostate as well as identifying potential therapeutic targets for future studies.
Similar content being viewed by others


Main
Diabetes mellitus (DM) is a common disease worldwide. Its prevalence has been increasing dramatically in recent years.1, 2 Diabetics mellitus is characterized by an increased concentration of glucose in the blood due to insulin deficiency or decreased tissue response to insulin, resulting in metabolic alterations of protein, carbohydrate and lipid in many important organ systems.
In alloxan-induced diabetic rats, Arcolino et al3 found that soon after the onset of diabetes, there are important changes in cell proliferation within the prostatic epithelium, and in longer term, there is a marked impact on the kinetics of differentiation and cell death. Soudamani et al4 found that the diabetic rats showed disturbed development and differentiation of the ventral lobe of the prostate. Histologically, glandular epithelium undergoes shrinkage with transformation of acinar cells into low cuboidal type with less prominent secretory granules. Interstitial space is increased owing to shrinkage of glandular epithelium. Histomorphometric studies on the tubular diameter, volume and surface density of acinar epithelium, lumen and stroma also support regressive changes in the prostate. Suthagar et al5 reported adverse effects of streptozotocin (STZ)-induced diabetes on the biochemical profiles of rat ventral prostate. Glucose oxidation, androgen and estrogen receptor concentration were also decreased in ventral prostate. Specific activities of both acid and alkaline phosphatases were also markedly decreased owing to diabetes. Amelioration of these changes (partially or completely) by insulin replacement indicates that optimal insulin is essential for maintaining functional integrity of ventral prostate.
Although the previous studies have established important link between diabetes and prostate pathology, the molecular mechanisms remain unknown. To address this issue, we used cDNA microarray to analyze a STZ-induced diabetic rat model. Some results were confirmed by real-time polymerase chain reaction (PCR) and immunohistochemistry staining of the rat and human prostate tissue samples. An in vitro cell culture model was used to study the direct effect of high concentration of glucose. Our study offers important insights into understanding the molecular pathogenesis of the diabetic prostate and identifies potential novel targets for therapies.
MATERIALS AND METHODS
Experimental Animal Model
Experimental procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, revised 1996) and were performed according to the institutional ethical guidelines for animal experiments. In all, 20 male 12-week-old SD rats (weight 230–280 g), obtained from Sun Yat-sen University Experimental Animal Center (Guangzhou, China), were randomly divided into the diabetes and the control groups, with 10 rats in each group. The rats were housed in polycarbonate cages in a controlled environment, with an illumination schedule of 12 h light/12 h dark. Diabetic condition was induced by a single intraperitoneal injection of STZ (60 mg/kg (Sigma), dissolved in an ice-cold 0.1 M citrate buffer). STZ is a widely used N-nitroso derivative of D-glucosamine, which functions as a toxin that selectively destroys the insulin-producing cells of the pancreas, rendering the rat diabetic within 24 h of injection.5, 6 Blood glucose levels were monitored to confirm the presence of diabetes (blood glucose >300 mg/dl). Control rats were injected with citrate buffers only. After 4 weeks, rats in both groups were killed by pentobarbital (60 mg, intraperitoneal). The ventral lobe of the prostate from each rat was dissected immediately. Each tissue block was cut into two parts, with one-half used for pathological examination (morphological and immunohistochemical analysis) and the other half for molecular analysis (microarray and real-time PCR detection), which were kept frozen at −80°C until they were microdissected.
Morphological Analysis
Samples obtained from diabetic rats and control rats were submitted for morphological analysis. Paraffin-embedded tissue samples were cut into 5-μm sections, stained with hematoxylin–eosin (H&E). For ultrastructural studies, samples were fixed with 4% paraformaldehyde for 2 h, followed by 1 h in 1% osmium tetroxide in cacodylate buffer. The materials were embedded in resin, cut into 0.5-μm-thick sections and mounted on copper grids. The sections were then stained with uranyl acetate and lead citrate, examined and photographed with an electron microscope as described previously.7
cDNA Microarray Data Analysis
Total RNAs from the ventral lobe of the prostate were harvested by using TRIzol (Invitrogen) and the RNeasy kit (Qiagen) according to the manufacturer's instructions, including a DNase digestion step. RNA quality was assessed using a spectro-photometer (NanoDrop; Lab tech International, Ringmer, UK) and a Bioanalyzer 2100 (Agilent, South Queensferry, UK). RNA was generally of high quality (average 260/280, ratio 2:14). The samples were amplified and labeled using the Agilent Quick Amp labeling kit. The labeled probes were then hybridized with the Whole Rat Genome Oligo Microarray Kit (including 41 000 rat genes and transcripts; Agilent) in the hybridization buffer for 17 h at 65°C.
After hybridization, the slides were washed in 0.5 × SSC/0.01% SDS for 5 min and 0.06 × SSC for 2 min at room temperature. The Cy3 and Cy5 fluorescent intensities for each spot were determined by an Agilent DNA Microarray Scanner (performed by KangChen Bio-tech, Shanghai, China).
The resulting text files extracted from the Agilent Feature Extraction Software (version 10.5.1.1) were imported into the Agilent GeneSpring GX software (version 10.0) for further analysis. The microarray data sets were normalized in GeneSpring GX using the Agilent FE one-color scenario (mainly median normalization). The positive effect of this median normalization is illustrated in box-plot. Differentially expressed genes were identified through fold-change screening.
Three normalization steps were applied as follows: data transformation, dye swap; per spot, divide by control channel; and per chip, normalization to the 50th percentile. Genes were filtered in accordance with the following three criteria: an average raw and control value 100 in all chips; a present or marginal flag value for both samples in each tissue chip; a percent difference between raw and control values within 51% in each tissue chip. Genes that met all criteria were extracted. Data analysis was focused on genes whose levels of expression increased or decreased by twofold or greater in diabetes rats compared with the control rats. GO analysis and Pathway Analysis were performed on this subset of genes. The function of extracted genes was sorted based on the analytical program of Database for Annotation Visualization and Integrated Discovery.8
Real-Time PCR
Real-time PCR was performed as described previously.9 Briefly, the total RNA extracted from the same RNA pool of microarray was used as the starting material for reverse transcription by using the Moloney murine leukemia virus reverse transcriptase system (Invitrogen). Real-time PCR was carried out with SYBR Green PCR Master Mix (Bio-Rad, Hercules, CA, USA). A total of 40 cycles of PCR reactions were performed with 30 s of denaturing at 95°C, 30 s of annealing at 60°C and 1 min of PCR reaction at 72°C on an iCycler iQ real-time PCR system (Bio-Rad). β-Actin was used as the control. The primer sequences used are described in Table 1. To confirm the changes of gene expression, all samples were first normalized to β-actin and the relative normalized units were compared between the diabetic group and control samples. Fold changes in expression were calculated using the 2−ΔΔCt method by an iCycler iQ software (Bio-Rad). The experiment was carried out in triplicate.
Human Prostate Tissue Samples
All tissues were obtained with the informed consent of each patient and in accordance with approved protocols and guidelines of our institutional review board. Prostate tissue samples of 25 patients with a history of more than 10 years of diabetes accompanied with benign prostatic hyperplasia (BPH) were collected during the surgery of transurethral resection of the prostate. Prostate tissue samples from 25 cases of age-matched BPH patients without diabetes were used as controls. The prostatic tissues were embedded in paraffin. Sections of 5 μm thickness were prepared and used for immunohistochemistry.
Immunohistochemical Staining
Deparaffinized tissue sections were subjected to antigen retrieval in citrate buffer pH 6.0, microwave at 750 W for 20 min, followed by treatment for 10 min in 3% (v/v) H2O2 in PBS to block endogenous peroxidases. They were subsequently incubated in blocking solution for 10 min to eliminate nonspecific bindings. Serum was removed from the slides and the slides were then incubated with the primary antibodies including rabbit anti-human and rodent Xrcc3 (no. X4003; Sigma), rabbit anti-rodent Xrcc2 (no. X3878; Sigma) and rabbit anti-human and rodent Mre11 (no. 4895; Cell Signaling Technology) (all diluted in 1:100), respectively, at 4°C overnight. Following incubation, the slides were washed three times in PBS for 3 min each. The slides were then incubated with a secondary antibody (polyclonal donkey anti-goat or goat anti-rabbit antibody; Santa Cruz) for 30 min at RT, followed by three washes in PBS for 3 min each. The slides were then incubated with Strept ABC Complex/HRP (Dako) for 30 min in a humidified chamber at RT, followed by three washes in PBS for 3 min each. A 3,3′diaminobenzidine substrate (Vector Laboratories) was used for detection and hematoxylin was used for counterstaining. The slides were then dehydrated, mounted and scanned. Negative controls were obtained by omission of the primary antibody. Immunostaining intensity was scored as reported previously.9 Briefly, protein expression was scored using two measures: intensity on a 0–3 scale (0=negative; 3=strongly positive) and percentage of positively stained cells (range 0–100% positive) at each intensity scale. An integrated intensity score was calculated by using the formula: ((% staining at intensity 3) × 3+(% staining at intensity 2) × 2+(% staining at intensity 1) × 1)/100 as reported previously.10
Cell Culture
The normal human prostatic epithelial cell line, RWPE-1, obtained from American Type Culture Collection, was maintained in K-SFM medium (Invitrogen) containing 50 mg/ml bovine pituitary extract, 5 ng/ml epidermal growth factor and 1 × antibiotic/anti-mycotic solution (Invitrogen). N-acetyl-L-cysteine (NAC; Sigma), acting as a reactive oxygen species (ROS) scavenger in this study, was bought from Sigma. For high glucose exposure, cells were maintained in the medium containing 25–75 mM glucose for various time points, with or without NAC pretreatment for 30 min11, 12
Western Blot Analysis
Cell lysates were centrifuged at 14 000 g for 30 min at 4°C. Equal amounts of cellular protein, as determined by Bradford dye-binding assay (Bio-Rad), were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto 0.45-μm (pore size) nitrocellulose. After blocking with 5% non-fat dry milk for 1 h at room temperature, membranes were probed with the primary antibodies: rabbit anti-human Xrcc3 (no. X4003; Sigma) and rabbit anti-human Mre11 (no. 4895; Cell Signaling Technology) (both diluted in 1:1000), respectively, for 16 h at 4°C, and then with secondary antibody. Bands were visualized with the Enhanced Chemiluminescence System (GE Life Science), and band density was detected with the FUJI film LAS-3000 system and quantified.9 β-Actin was used as the loading control.
Assessment of Intracellular ROS
The levels of ROS generated in RWPE-1 cells were measured using 2,7-dichlorodihydrofluorescindiacetate (DCFH-DA) as a fluorescence probe. RWPE1 cells were serum-deprived for 24 h, and incubated for 30 min at 37°C with 10 μmol/l of DCFH-DA (Molecular Probes).12 Cultures were then exposed to high glucose (25–75 mmol/l) for various time courses. Cells were harvested, and washed three times with PBS. The photos of DCF-DA fluorescence were immediately recorded. Cells treated identically were also subjected to analysis by a flow cytometer (Becton-Dickinson) at an excitation wave length of 485 nm and emission wave length of 530 nm.12
Analysis of 8-OHdG in the Prostate Tissues
The levels of 8-OHdG in the rat prostate tissues were measured by using an enzyme-linked immunosorbent assay kit as described previously.13, 14 Prostate DNA was isolated and digested with DNase for 10 min, and transferred to the pre-coated 8-OHdG microtiter plate, and incubated with a monoclonal antibody against 8-OHdG at 37°C for 1 h. After washing, samples were incubated with the secondary antibody at 37°C for 1 h, followed by treatment with substrate solution. The absorbance was measured at 450 nm using the microtiter plate reader.
Statistical Analysis
Data were presented as the means±s.e. Statistical comparisons between groups were performed by t-test and P<0.05 was considered to be significant.
RESULTS
Changes in Body and the Organ Weight after Diabetes
STZ-induced diabetes caused a significant decrease in body weight of the rats (control vs diabetes, 426.3±24.8 vs 315.6±20.5 g) and the relative weight of the ventral prostate (1.2±0.13 × 10−3 vs 0.86±0.07 × 10−3), as well as a significant increase in the blood glucose levels (87.5±7.6 vs 423.4±12.4 mg/dl).
Light Microscopic and Electron Microscopic Findings
Histological examination of H&E-stained tissue sections under light microscope showed regressive morphological changes in the prostate induced by diabetes, compared with the control group (Figure 1a and c), which was consistent with the previous reports. In the diabetic group, mucosal folds were decreased and the secretary epithelium was atrophic (Figure 1b and d). The epithelial cells displayed decreased total cell volume with nuclei occupying a larger area of the cells. The stroma was thin with smooth muscle fibers and fibroblasts as the main components.

Morphology of the prostates from control and diabetic rats. (a) Control, × 200 and (c) control, 00 are light photomicrographs from control prostates showing large acini with columnar epithelium. (b) Diabetic, × 200 and (d) diabetic, × 400 are light photomicrographs from the diabetic group showing atrophic secretary epithelium with small nuclei and scanty cytoplasm. Electron photomicrographs from the control (e, × 2200; g, × 15 500) and the diabetic prostates (f, × 2200; h × 15 500). Compared with the control group, the diabetic group showed reduction in glandular epithelium cell height (f), discontinuous microvilli and fewer secretary vacuoles in the apical region (h). (Arrows represent the height of the epithelial cells. N, nuclears; M, microvilli; SG, secretory granule.)
Examination under electron microscope showed reduction in the height of the glandular epithelial cells, resulting in cuboidal shapes of these cells (Figure 1f). The apical region of the diabetic prostates had decreased amount of secretion, reduced numbers and discontinuous arrangement of microvilli, as well as reduced secretary vacuoles (Figure 1f and h) in comparison to the control prostates (Figure 1e and g).
cDNA Microarray Analysis
To identify the underlying molecular changes of diabetic prostate, cDNA microarray analysis was carried out. Nearly 41 000 transcripts were detected in both groups. Among them, 2304 genes displayed a more than twofold changes in expression levels in the diabetic group, as compared with the control group. A partial list is shown in Tables 2 and 3. The altered genes can be categorized as (1) proliferation and differentiation related, (2) anti-oxidant enzymes, (3) DNA damage repair genes, (4) cell cycle checkpoints related, (5) angiogenesis related and (6) apoptosis regulation related. Other major clusters observed are those involved in signal transduction, cholesterol metabolism, fatty acid biosynthesis, lipid and insulin metabolism, among others (Figure 2).

Major clusters of altered genes identified in the complementary DNA (cDNA) microarray analysis. P<0.05.
Confirmation of Gene Expression Profile Results by Real-time PCR Analysis
In all, 13 genes from the microarray study were chosen for real-time PCR analysis on the basis of their functions in DNA damage repair (Rad51L3, Xrcc3, Xrcc2, Mre11), cell cycle checkpoints (Ccna2, Ccne1, Tcfdp2, Mcm8), proliferation (Cdkn2a) and apoptosis (Mapt, Dffa, Vim, Caspase 9). As shown in Figure 3, the results of real-time PCR analysis were consistent with those of microarray analysis.

Confirmation of altered gene expression by real-time polymerase chain reaction (PCR) analysis. Relative quantification of selected genes from the control and diabetic groups was calculated by normalizing individual gene expression to the housekeeping gene, β-actin. The subtable indicates the covalence of gene expression between the quantitative PCR and microarray analysis (P<0.05).
Confirmation of Gene Expression Profile Results by Immunohistochemical Staining
Consistent with the results of microarray and real-time PCR, expression of MRE11 and XRCC3, not XRCC2, were markedly downregulated in the diabetic prostate (Figure 4a). Furthermore, we found that both XRCC3 and MRE11 were also decreased in the human prostate tissues from clinical patients with more than a 10-year history of diabetes, as compared with those of the control group (Figure 4b). These results suggest that diabetes induces a significant change in the expression of DNA repair genes.

The expression of selected proteins in the prostate tissues. (a) Immunohistochemical staining of MRE11, XRCC3 and XRCC2 in the rat prostate tissues. (b) Immunohistochemical staining of MRE11 and XRCC3 in prostate tissues of benign prostatic hyperplasia (BPH) patient samples (P<0.05).
High Glucose Increased ROS in Prostate Cells
To study the direct effect of high glucose, an in vitro cells’ culture model was used. Exposure of human prostate epithelial cells RWPE-1 to a high concentration of glucose (75 mM) for 15 min to 72 h elicited a significant increase of intracellular ROS level. DCF fluorescence reflecting the relative levels of ROS was shown in Figure 5a. This finding was confirmed by flow cytometric analysis (Figure 5b). Figure 5c shows a dose–response relationship between glucose levels and DCF fluorescence (the ROS indicator). These results indicate that high glucose can directly induce oxidative stress in the prostate.

The generation of reactive oxygen species (ROS) in cultured RWPE-1 cells exposed to high glucose (HG). Serum-starved (0.5% fetal bovine serum (FBS)) cells were exposed to HG. At indicated time points, cells were washed and loaded with 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) to capture ROS. (a) Images taken by a fluorescence microscope. The intensity of fluorescence was measured by using the software of Image J. The figures are representative of three independent experiments. (b) The fluorescence of DCF induced by HG (75 mM) for 15 min and 48 h were detected by flow cytometry as described in Materials and Methods. (c) Treatment of HG for 48 h induced increase of DCF fluorescence in a dose-dependent manner. The data (mean fluorescence in each group) was presented as bar charts (error bars indicate s.d. of triplicate experiments).
Increased Levels of DNA Damage in the Diabetic Group
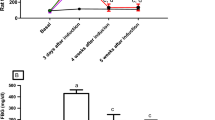
8-OHdG is a specific indicator of oxidative DNA damage in the tissues.13 As shown in Figure 6, the level of 8-OHdG was significantly increased in the diabetic group compared with the controls, suggesting the presence of oxidative DNA damage in the diabetic prostate tissues.

Enzyme-linked immunosorbent assay (ELISA) assay showing increased levels of DNA damage marker 8-hydroxy-2′-deoxyguanosine (8-OHdG) in the prostate tissues of the diabetic rats.
High Glucose Downregulates the Expression of DNA Repair Genes
In cultured RWPE-1 cells, exposure to high concentration of glucose, not to the mannitol control, caused decreased expression of MRE11 and XRCC3 in a dose- and time-dependent manner (Figure 7). This phenomenon could be reversed by the anti-oxidant NAC, a scavenger of ROS, indicating that oxidative stress was involved in altered expression of DNA repair genes by high concentrations of glucose.

Effects of high glucose (HG) on XRCC3 and MRE11 expression in RWPE-1 cells. β-Actin was used as the loading control. (a) Cells were treated with different concentrations of glucose as indicated. Mannitol (75 mM) was used as the osmetic control. (b) Cells treated with HG (75 mM) shown at different times. HG decreased the expression of MRE11 and XRCC3. This effect was blocked if cells were pretreated with ROS scavenger N-acetyl-L-cysteine (NAC) for 30 min. Results were representative of three independent experiments. Right panel shows densitometry analysis of protein levels. The density readings for MRE11 and XRCC3 were standardized to those of β-actin. Bars, s.d. P<0.05 by unpaired t test. (NG, normal glucose (5.5 mM).)
DISCUSSION
In this study, we performed whole-genome analysis of the prostate in diabetic conditions, and confirmed the changes of some new targets in both rat and human prostate tissues. Diabetic conditions in our experimental model induce changes in the expression of gene associated with many important cellular functions. One caveat of the study is that the diabetic condition was induced by STZ; hence, the possibility of a direct effect of STZ on these genes cannot be entirely ruled out, although our confirmatory studies of a subset of the genes in human prostate and cell culture strongly support a direct relationship between diabetes and the gene expression changes. It must also be noted that diabetes is more than hyperplasia as it induces changes in many other physiological processes in multiple organs including significant changes in many hormones. Another point worthy of discussion is that our control human material is from patients with BPH, which is not considered normal prostate tissue. However, as both BPH and prostate cancer are diseases of older men, cancer often arises in prostate gland with BPH. Therefore, BPH tissue is an appropriate control for prostate cancer.
Diabetes causes many complications in multiple organs and may be related to cancer risks.15 Most available clinical studies showed that diabetes decreases the risk of prostate cancer in general. It appears to decrease mainly the risk of low-grade prostate cancer, and to a lesser degree that of high-grade prostate cancer.15, 16, 17 However, different findings have also been reported. Recent publications showed that it may actually increase the risk of prostate cancer18 and prostate cancer mortality19 in Taiwanese population, who should have similar or identical genetic background to the patient population in our study. It is uncertain if the discordant findings may represent possible racial differences, a topic that warrants additional large-scale studies.
Diabetes can promote oxidative stress in some organs.20, 21 In this study, we found that high glucose causes direct oxidative stress and disruption of intracellular redox homeostasis in the prostate. Direct exposure of human prostate epithelial cells (RWPE1) to high glucose increases the generation of intracellular ROS. Meanwhile, we noticed that many anti-oxidant enzymes, such as Gpx1, Gpx, Oplah, Gstm4, Mgst2, Cyp4b1 and Dpyd, were all significantly changed in the diabetic group. Accordingly, 8-OHdG, a specific marker of oxidative DNA damage,14 was significantly increased in the tissue of the diabetic prostate, as compared with the control.
ROS plays a vital role in the regulation of important cellular processes such as proliferation, apoptosis, and senescence.22 Excessive production of ROS or inadequacy in a normal cell’s anti-oxidant defense system (eg, anti-oxidant enzymes), can cause the cells to experience oxidative stress. Increased ROS generation has been associated with tissue injury or DNA damage which are general manifestations of pathological conditions associated with aberrant growth and neoplastic transformation.23
In this study, we found that many DNA damage repair genes, such as Mre11, Xrcc3, Rad51L3, dpoLL, Nudt5, Msh5, Msh3, Ube2c and SEMA6c, were all downregulated in the diabetic prostate, indicating significant defects in DNA repair machinery in the diabetic condition. MRE11 was reported to activate the ataxia–telangiectasia-mutated kinase1 and trigger DNA damage repair reactions.24, 25 XRCC3, a new Rad51-family member, is able to promote the repair of DNA damage and maintain the stability of the chromosome.26, 27, 28 We showed that expression of both MRE11 and XRCC3, not XRCC2, were decreased in the diabetic prostate. Moreover, the effect of high glucose on these two genes could be partly reversed by a ROS scavenger (NAC), suggesting that MRE11 and XRCC3 are the downstream effectors of ROS signaling in the prostate induced by high glucose.
Genomes are often exposed to a variety of DNA-damaging agents that cause DNA double-strand breaks, which can lead to cell death or trigger tumorigenesis. Normal cells have an efficient DNA repair machinery to cope with DNA damages.24, 25 However, once the function of this vital machinery is compromised, cells cannot maintain genetic stability and may result in neoplastic transformation. Epidemiological data suggest that diabetes mellitus is associated with increased risk of numerous cancers including those of the pancreas, liver, esophagus, colon/rectum, kidney, endometrium and breast, indicating that diabetes produces detrimental molecular changes in multiple organs,29 but the underlying molecular mechanism is poorly understood. So far, epidemiological studies investigating the association between diabetes and prostate cancer have achieved inconsistent results.30, 31, 32 Some studies suggested an inverse correlation, whereas others have indicated a positive correlation. Our findings suggest that diabetes may likely change prostate cell’s DNA repair machinery and result in increased susceptibility to malignant transformation. However, more studies are needed to draw definitive conclusions.
It has been reported that ROS is inherent in prostate cancer.33 Based on our findings that high concentration of glucose causes increased intracellular oxidative stress, DNA damage, deficiency of anti-oxidative enzymes and DNA damage repair machinery in the prostate, it can be speculated that restoration of DNA repair machinery may help to protect prostate cells from the oxidative DNA damage induced by high glucose, and could be developed as a potential strategy to decrease the risk of prostate cancer associated with diabetes.
Diabetes causes multiple genetic changes and produces a negative impact such as oxidative stress and defects of anti-oxidant DNA damage repair in the prostate. Our findings offer important clues for understanding the molecular pathogenesis of the diabetic prostate and suggest novel targets for potential therapies.
References
Yang WM, Lu JM, Weng JM, et al. Prevalence of diabetes among men and women in China. N Engl J Med 2010;362:1090–1101.
Weng J, Li Y, Xu W, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet 2008;371:1753–1760.
Arcolino FO, Ribeiro DL, Gobbo MG, et al. Proliferation and apoptotic rates and increased frequency of p63-positive cells in the prostate acinar epithelium of alloxan-induced diabetic rats. Int J Exp Pathol 2010;91:144–154.
Soudamani S, Yuvaraj S, Malini T, et al. Experimental diabetes has adverse effects on the differentiation of ventral prostate during sexual maturation of rats. Anat Rec A Discov Mol Cell Evol Biol 2005;287:1281–1289.
Suthagar E, Soudamani S, Yuvaraj S, et al. Effects of streptozotocin (STZ)-induced diabetes and insulin replacement on rat ventral prostate. Biomed Pharmacother 2009;63:43–50.
Mitsuma T, Nogimori T . Effects of streptozotocin-induced diabetes mellitus on hypothalamic-pituitary-thyroid axis in rats. Endocrinol Jpn 1982;29:695–700.
Favaro WJ, Padovani CR, Cagnon VH . Ultrastructural and proliferative features of the ventral lobe of the prostate in non-obese diabetic mice (NOD) following androgen and estrogen replacement associated to insulin therapy. Tissue Cell 2009;41:119–132.
Dennis GJ, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:P3.
Wen X, Li X, Liao B, et al. Knockdown of p21-activated kinase 6 inhibits prostate cancer growth and enhances chemosensitivity to docetaxel. Urology 2009;73:1407–1411.
Wen XQ, Li XJ, Su ZL, et al. Reduced expression of alpha-tocopherol-associated protein is associated with tumor cell proliferation and the increased risk of prostate cancer recurrence. Asian J Androl 2007;9:206–212.
Sharifi AM, Eslami H, Larijani B, et al. Involvement of caspase-8, -9, and -3 in high glucose-induced apoptosis in PC12 cells. Neurosci Lett 2009;459:47–51.
Samikkannu T, Thomas JJ, Bhat GJ, et al. Acute effect of high glucose on long-term cell growth: a role for transient glucose increase in proximal tubule cell injury. Am J Physiol Renal Physiol 2006;291:162–175.
Shimoi K, Kasai H, Yokota N, et al. Comparison between high-performance liquid chromatography and enzyme-linked immunosorbent assay for the determination of 8-hydroxy-2′-deoxyguanosine in human urine. Cancer Epidemiol Biomarkers Prev 2002;11:767–770.
Wu LL, Chiou CC, Chang PY, et al. Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin Chim Acta 2004;339:1–9.
Leitzmann MF, Ahn J, Albanes D, et al. Diabetes mellitus and prostate cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Cancer Causes Control 2008;19:1267–1276.
Gong Z, Neuhouser ML, Goodman PJ, et al. Obesity, diabetes, and risk of prostate cancer: results from the prostate cancer prevention trial. Cancer Epidemiol Biomarkers Prev 2006;15:1977–1983.
Abdollah F, Briganti A, Suardi N, et al. Does diabetes mellitus increase the risk of high-grade prostate cancer in patients undergoing radical prostatectomy? Prostate Cancer Prostatic Dis 2011;14:74–78.
Tseng CH . Diabetes and risk of prostate cancer: a study using the National Health Insurance. Diabetes Care 2011;34:616–621.
Tseng CH . Prostate cancer mortality in Taiwanese men: increasing age-standardized trend in general population and increased risk in diabetic men. Ann Med 2011;43:142–150.
Brownlee M . Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813–820.
Robertson RP, Harmon J, Tran PO, et al. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003;52:581–587.
Lau AT, Wang Y, Chiu JF . Reactive oxygen species: current knowledge and applications in cancer research and therapeutic. J Cell Biochem 2008;104:657–667.
Cooke MS, Evans MD, Dizdaroglu M, et al. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003;17:1195–1214.
Czornak K, Chughtai S, Chrzanowska KH . Mystery of DNA repair: the role of the MRN complex and ATM kinase in DNA damage repair. J Appl Genet 2008;49:383–396.
Zha S, Boboila C, Alt FW . Mre11: roles in DNA repair beyond homologous recombination. Nat Struct Mol Biol 2009;16:798–800.
Liu N, Lamerdin JE, Tebbs RS, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell 1998;1:783–793.
Cui X, Brenneman M, Meyne J, et al. The XRCC2 and XRCC3 repair genes are required for chromosome stability in mammalian cells. Mutat Res 1999;434:75–88.
Petermann E, Orta ML, Issaeva N, et al. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010;37:492–502.
Vigneri P, Frasca F, Sciacca L, et al. Diabetes and cancer. Endocr Relat Cancer 2009;16:1103–1123.
Snyder CF, Stein KB, Barone BB, et al. Does pre-existing diabetes affect prostate cancer prognosis? A systematic review. Prostate Cancer Prostatic Dis 2010;13:58–64.
Kasper JS, Giovannucci E . A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev 2006;15:2056–2062.
Tavani A, Gallus S, Bertuzzi M, et al. Diabetes mellitus and the risk of prostate cancer in Italy. Eur Urol 2005;47:313–317.
Kumar B, Koul S, Khandrika L, et al. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 2008;68:1777–1785.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants 30872585, 30901768 and 81072115), The Fundamental Research Funds for the Central Universities of China (Sun Yat-sen University, Grant 10ykpy06).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The molecular impact of diabetes mellitus on the prostate is elucidated using an STZ-induced diabetic rat model, human prostate tissue and cell culture studies. Diabetes has widespread effects on the expression of genes related to cell proliferation, oxidative stress, DNA damage repair, cell cycle regulation, angiogenesis and apoptosis.
Rights and permissions
About this article
Cite this article
Ye, C., Li, X., Wang, Y. et al. Diabetes causes multiple genetic alterations and downregulates expression of DNA repair genes in the prostate. Lab Invest 91, 1363–1374 (2011). https://doi.org/10.1038/labinvest.2011.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.87
Keywords
This article is cited by
-
High glucose promotes benign prostatic hyperplasia by downregulating PDK4 expression
Scientific Reports (2023)
-
Effects of maternal diabetes on male offspring: high cell proliferation and increased activity of MMP-2 in the ventral prostate
Cell and Tissue Research (2014)
