
Abstract
Critical limb ischemia causes severe damage to the skeletal muscle. This study develops a reproducible model of myotube ischemia by simulating, in vitro, the critical parameters that occur in skeletal muscle ischemia. Monolayers of C2C12 myoblasts were differentiated into mature myotubes and exposed to nutrition depletion, hypoxia and hypercapnia for variable time periods. A range of culture media and gas mixture combinations were used to obtain an optimum ischemic environment. Nuclear staining, cleaved caspase-3 and lactate dehydrogenase (LDH) release assay were used to assess apoptosis and myotube survival. HIF-1α concentration of cell lysates, pH of conditioned media as well as partial pressures of oxygen (PO2) and carbon dioxide (PCO2) in the media were used to confirm ischemic simulation. Culturing myotubes in depleted media, in a gas mixture containing 20% CO2+80% N2 for 6–12 h increased the PCO2 and decreased the pH and PO2 of culture media. This attempts to mimic the in vivo ischemic state of skeletal muscle. These conditions were used to study the potential tissue-protective effects of erythropoietin (EPO) in C2C12 myotubes exposed to ischemia. EPO (60 ng/ml) suppressed LDH release, decreased cleaved caspase-3 and reduced the number of apoptotic nuclei, suggesting significantly decreased ischemia-induced apoptosis in myotubes (P<0.01) and a potential role in tissue protection. Additional therapeutic agents designed for tissue protection can also be evaluated using this model.
Similar content being viewed by others
Main
Critical limb ischemia (CLI), a severe form of peripheral arterial disease, causes severe morbidity and mortality. It affects between 500 and 1000 individuals per million per year.1 One-year amputation and mortality rates in this subgroup are 30 and 25%, respectively,2 and current pharmacotherapy options are limited.3 Therefore, there is potential to develop better therapeutic agents for treatment of CLI.
CLI causes apoptosis of skeletal muscle that forms the bulk of the lower extremity.4, 5 Some other important changes seen in skeletal muscle ischemia in vivo are listed in Table 1. Here, we characterize and simulate conditions in vitro that may bear resemblance to CLI.
In vitro models are useful for the initial assessment of new formulations of existing compounds as well as novel agents. Although simulated ischemia models have been widely used for cardiomyocytes6, 7 and neurons,8, 9 an efficient reproducible model has not been optimized for skeletal muscle ischemia. Such a model would help in the investigation of compounds aimed at tissue protection and to identify important regulatory signaling and transcription pathways.
Skeletal muscle tissue contains various cell types including adipocytes, fibroblasts, nerve cells and stromal vascular components. However, myotubes predominantly influence the local milieu.10, 11 Therefore, a model based on myotubes was developed using differentiated C2C12 myoblasts.12 This skeletal muscle-like cell line is well characterized,12 commercially available, easy to culture and has been used as a myogenesis model in unrelated studies.13, 14, 15
Erythropoietin (EPO) has been traditionally used to treat anemia resulting from chronic renal failure.16 However, it is now well known that it has tissue-protective properties.17, 18, 19, 20 Furthermore, the authors have recently reported that EPO receptors (EpoR) are upregulated in human skeletal muscle obtained from patients with CLI,21 suggesting the potential of EPO and its derivatives in treating CLI. The possible effect of EPO on preventing apoptosis in skeletal muscle and the pathways involved, however, are unknown and can be assessed using this model.
The hypotheses of the study are: first, exposure of mature C2C12 myotubes to nutrition depletion, hypoxia and hypercapnia for a specific time period can simulate aspects of skeletal muscle ischemia, including pH, partial pressure of oxygen and carbon dioxide, as well as apoptosis of skeletal myotubes. Second, EPO decreases ischemia-induced apoptosis in a monolayer of myotubes.
METHODS
Cell Culture and Incubation Procedure
The C2C12 mouse myoblast cell line was obtained from the European Collection of Cell Cultures (ECACC no. 91031101, passage no. 13, Salisbury, UK). The myoblasts were grown in Dulbecco's modified Eagle's medium (DMEM; 1 × , liquid, high glucose, catalog number (cat. no.) 31966-021; Invitrogen, Paisley, UK). The culture medium was supplemented with 10% fetal calf serum (GIBCO®, Paisley, UK), 100 U/ml penicillin and 100 g/ml streptomycin. The incubation was performed in humidified atmosphere of 21% O2, 74% N2 and 5% CO2 at 37°C. The myoblasts were grown in a T75 cell culture flask (BD Falcon™, Franklin Lakes, NJ, USA) containing 25 ml of culture medium. The media were changed every 48 h. Myoblasts were cultured to a maximum of 50% confluence at this stage, to avoid contact inhibition of mitosis. Cells between 3 and 12 passages were used. After trypsinization and re-suspension of adherent cells, cell density was assessed by a hemocytometer. A seeding density between 10 000 and 50 000 cells per ml of culture media was used for plating the culture dish for experiments. A variety of culture dishes were used to enable staining and microscopy as well as with collection of cell lysates for analyses. The volume of culture media used in each was as follows:
-
12-well culture plate (BD Falcon™, cat. no. 351143)=2 ml per well.
-
35 × 10 mm2, Easy-Grip™ Cell Culture Dish (BD Falcon™, cat. no. 353001)=2.5 ml per well.
-
4-well culture slide (BD Falcon™, cat. no. 354104)=1 ml per well.
Myoblasts were then cultured to 80% confluence, after which fetal calf serum was replaced with 2% heat-inactivated horse serum to prevent myoblasts from dividing further and to ensure maturation into myotubes. The culture media were changed daily until the myotubes appeared to be fully mature, which usually took 1 week. Compounds being tested were added to the culture dish 24 h before subjecting cells to simulated ischemia.
Simulated Ischemia
Ischemia constitutes metabolic waste accumulation and nutrition depletion in addition to hypoxia.22 Therefore, to simulate nutrient depletion and waste accumulation, the culture media were either left unchanged (stagnant culture media) or replaced with serum-deprived low glucose (Invitrogen, cat. no. 21885108) and glucose-free DMEM (Invitrogen, cat. no. 11966-025) to determine if better ischemic simulation was achieved with stagnant culture media in comparison to depleted fresh medium. The cells (in open dishes) were then exposed to gas mixture-1 (20% CO2+80% N2) or gas mixture-2 (5% CO2+94% N2+1% O2) obtained from pre-mixed gas tanks (British Oxygen, Luton, UK) in hypoxic chambers (Modular Incubator Chamber, MIC-101; Billups-Rothenberg, Del Mar, CA, USA). The chamber was flushed with gas at 10 l/min for 20 min and then sealed. The apparatus was placed in an incubator at 37°C for 4, 6, 8 and 12 h. Control myotubes were maintained under normoxic conditions (21% O2+5% CO2) for equivalent periods of time with standard culture media changes. Gas analysis of 1 ml of conditioned media was performed immediately at release of the gas chamber after 4, 6, 8 and 12 h using an automated blood-gas analyzer (Rapidlab 800 series; Bayers Healthcare LLC, East Walpole, MA, USA). These allowed partial pressures of O2 and CO2 along with pH of the various conditioned media to be obtained after different periods of hypoxia and that from control myotubes to be compared. The remaining 1 ml of medium was collected and clarified by centrifuging at 1600 g for 5 min at 4°C and stored at −80°C for the analysis of lactate dehydrogenase (LDH) release.
Cell Morphology and Viability
Cell viability was assessed both qualitatively and quantitatively. Myotube monolayers incubated under normal conditions and those exposed to simulated ischemia were stained with hematoxylin and eosin and examined under light microscopy for morphological changes associated with apoptosis. In addition, fluorescent nuclear staining to detect the number of apoptotic nuclei was performed as follows: VECTASHIELD® mounting medium containing 4′, 6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei of myotubes pre-fixed with 4% formalin solution. The morphology of cell nuclei was observed using a fluorescence microscope (Axioscop 2; Carl Zeiss Micro-imaging GmbH, Jena, Germany) at an excitation wavelength of 350 nm. Features of apoptotic cells included brightly stained nuclei with condensed chromatin drawn together at the fringes of the nuclear membrane or an entirely fragmented nucleus. Two blinded observers counted a minimum of three randomly chosen fields, of at least three independent experiments, per experimental condition. At least 250 individual nuclei were counted at one time. Data were expressed as the percentage of apoptotic cells and was statistically analyzed for significant difference between normoxia and ischemia as well as between different time periods of ischemia.
Immunofluorescence
After removal of chamber slides from the hypoxic chamber, the myotube monolayer was washed three times with phosphate-buffered saline (PBS) and fixed in 4% formaldehyde for 5 min. Serum block was performed with 10% goat serum for 20 min. Cleaved caspase-3 (Asp175) antibody (Cell Signaling Technology, Danvers, MA, USA), in a dilution of 1:50 and EpoR M-20 antibody (sc-697; Santa Cruz Biotechnology, Santa Cruz, CA, USA) in a dilution of 1:100 were used to probe the expression of cleaved caspase-3 enzyme and EpoR, respectively. After incubation with primary antibody or its IgG isotype control for 1 h at room temperature, the slides were washed with PBS and treated with Texas Red®-X goat anti-mouse IgG antibody (Invitrogen), in 1:1000 dilution for 60 min in the dark at room temperature. The slides were washed with PBS and coverslips were mounted using VECTASHIELD® mounting medium containing DAPI. The images were viewed using a fluorescence microscope (Axioscop 2; Carl Zeiss Micro-imaging).
Western Blotting
The myotube monolayer was harvested on ice, thoroughly washed with cold PBS and lysed in 75 μl RIPA buffer 1 × solution (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0; product number R0278; Sigma Aldrich, St Louis, MO, USA). Following this, 75 μl of Laemmli buffer was added to each well of the 12-well culture plate. The monolayer was scrapped with a cell scrapper and collected into a 1.5 ml Eppendorf tube using a syringe with a 23 G needle. A total of 5 μl (2–5%) of 2-mercaptoethanol was added to the cell extract, which was then heated at 95°C for 5 min to denature the proteins. Extracts were then centrifuged at 1600 g for 5 min at 4°C. The supernatant was collected in clean Eppendorf tubes and stored at −80°C for subsequent use in western blot analysis.
A total of 12 μl of sample was loaded onto each well of a 12% Tris-glycine gel (Invitrogen) and run with a broad-range protein marker. After transfer onto a nitrocellulose membrane (Hybond-C extra; GE Healthcare Life Sciences, Buckinghamshire, UK), nonspecific proteins on the membrane were blocked with PBS-Tween containing 5% milk for 2 h. The membranes were incubated overnight at 4°C with the following specific antibodies (dilution of 1:1000): cleaved caspase-3 (Asp175) antibody (Cell Signaling Technology); HIF-1α antibody (AF1935, R&D Systems, Minneapolis, MN, USA); myogenin antibody (sc-52903; Santa Cruz Biotechnology); EpoR (M-20) antibody (sc-697; Santa Cruz Biotechnology); and α-tubulin antibody—loading control (ab4074; Abcam, Cambridge, MA, USA). The membranes were then washed and incubated with horseradish peroxidase-linked corresponding secondary antibody at room temperature. The membrane was developed using chemiluminescent substrate (Amersham ECL Plus™ Western Blotting Detection Reagent; GE Healthcare Life Sciences) and the blots were developed against photographic film (Amersham Hyperfilm ECL, GE Healthcare Life Sciences). Band intensities were determined by densitometry using Biospectrum® AC imaging system (UVP, Upland, CA, USA).
LDH Assay
The LDH assay was performed according to the manufacturer's protocol. Fully automated ‘Cytotoxicity detection kit (LDH)’ (Roche/Hitachi 917-11876961 216, Mannheim, FR, Germany) was used to measure LDH release in units per liter (U/l) in culture media obtained from myotubes subjected to simulated ischemia for 4, 6, 8 and 12 h. The release of LDH into the culture medium was assayed by incubating the clarified culture medium with sodium pyruvate in the presence of NADH. Pyruvic acid is catalyzed into lactic acid by free LDH along with a simultaneous oxidation of NADH to NAD+. The rate of oxidation of NADH to NAD− was measured spectrophotometrically at 340 nm.
Pretreatment with EPO
Firstly, the expression of EpoR in C2C12 myotubes was shown by immunohistochemistry and western blot as described in the previous sections. Then, the simulated ischemia model was used to investigate the effect of EPO on myotubes exposed to ischemia. Differentiated C2C12 myotubes were cultured in culture dishes, as described in the previous section. They were treated with varying doses (1 ng/ml to 1 μg/ml) of recombinant human EPO (rhEPO, hBA-165: sc-4620; Santa Cruz Biotechnology), 24 h before exposure of the cells to simulated ischemia. The rationale for pre-treating the myotubes 24 h before they are subjected to ischemia is to allow activation of multiple antiapoptotic signaling pathways that mediate tissue protection. Compounds with rapid onsets of action may be added to the culture medium immediately before the onset of simulated ischemia. After pretreatment with rhEPO (with negative and positive controls), nuclear staining, LDH release assay and cleaved caspase-3 assay were performed to assess degrees of apoptosis (Supplementary Table online).
Statistics
All experiments were performed in triplicate and repeated at least three times. For quantitative variables, comparison was made between the mean±standard deviation of triplicate samples from the same experiment and from combined independent experiments, when inter-experiment variation allowed reliable combination of raw data. One-tailed Student's t-test was used to compare parametric data from two sets of independent samples and one-way ANOVA/Tukey's HSD test was used to compare parametric data from more than two sets of independent samples. Inter-observer variability was measured using linear correlation by calculating the Pearson's product–moment correlation coefficient (r). P-values <0.05 were considered significant. The data were analyzed using open-access online statistical computation software (http://faculty.vassar.edu/lowry/VassarStats.html).
RESULTS
Differentiation of C2C12 Myoblasts into Myotubes
Myoblasts differentiated into elongated, thickened and multinucleated myotubes after serum content of the culture media were reduced (Figure 1a–c). In addition to changes in morphology, quantifying the expression of myogenin protein provided further confirmation of differentiation, as myogenin is expressed preferentially in differentiated skeletal myotubes as opposed to myoblasts.23 Myogenin was significantly upregulated in myotubes as determined by western blotting in Figure 1d (P<0.0001).

Cell culture showing differentiation of myoblasts into myotubes. Hematoxylin and eosin (H&E) staining of C2C12 myoblasts (a) and myotubes (b). A magnified view of multinucleated myotubes (c). Representative western blot analysis of cell lysates obtained from monolayer of myoblasts and myotubes (day 7) shows a significant upregulation in the levels of myogenin in the myotubes (d). Data are represented by mean±s.d., n=9. P<0.001, one-tailed t-test for independent samples.
Hypoxia is Sustained During the Experiment
HIF-1α was significantly upregulated in the cell lysates obtained from myotubes subjected to simulated ischemia in gas mixture-1 for 8 h as determined by western blotting (P=0.01) shown in Figure 2. However, there was no significant difference in HIF-1α expression between gas mixture-1 and gas mixture-2 or between 6, 8 and 12 h of hypoxic exposure (data not shown).

Simulating hypoxia. Representative western blot analysis of hypoxia-inducible factor 1α (HIF-1α) in cell lysates obtained after exposure to simulated ischemia for 8 h. There was an upregulation of HIF-1α in ischemic cells in comparison to the ones incubated under normoxic conditions. Data are represented by mean±s.d., n=9. P=0.01, one-tailed t-test for independent samples.
The partial pressure of carbon dioxide (PCO2) (Figure 3a) was significantly increased in the conditioned media obtained after incubation in gas mixture-1 (P<0.01), but not in gas mixture-2 (P>0.05). However, the partial pressure of oxygen (PO2) (Figure 3b) was significantly lower in the conditioned media after incubation in both gas mixtures (P<0.01), with no significant difference between the PO2 of the conditioned media from gas mixture-1 and gas mixture-2 (P>0.05). In addition, there was no significant difference in the levels of PO2 and PCO2 at 4, 6, 8 and 12 h (data not shown). Figure 3 shows the data for PO2 and PCO2 of conditioned media obtained after 8 h of simulated ischemia.

Effect of simulated ischemia on partial pressure of oxygen (PO2), carbon dioxide (PCO2) and pH. There is a significant rise in the PCO2 (a), a significant fall in the PO2 (b) and a significant fall in the pH (c) in the conditioned media obtained after simulated ischemia. Gas mixture-1 containing 70% N2 and 30% CO2 was more effective than gas mixture-2 containing 94% N2, 4% CO2 and 1% O2. Data are represented by mean±s.d., n=9. One-way analysis of variance (ANOVA)/Tukey's HSD (honestly significant difference) test was used.
Simulated Ischemia Leads to a Fall in the pH of Culture Media
The mean pH for 0, 4, 6, 8 and 12 h of simulated ischemia was 7.43, 7.40, 7.28, 7.12 and 7.09, respectively. There was a significant fall in pH of the conditioned media (using gas mixture-1) between 6 and 12 h of ischemia. No significant fall in pH occurred when gas mixture-2 was used instead (P>0.05; Figure 3c). Hence, gas mixture-1 was more effective in reducing the pH of conditioned media.
Expression of EpoR
The expression of EpoR in C2C12 myotubes was confirmed before investigating the potential effects of EPO on ischemic C2C12 myotubes. A clear expression of the extracellular domain of EpoR in the myotube monolayer was noted on immunofluorescence (Figure 4a). The IgG isotype (negative control) showed no expression (Figure 4b). The expression was also shown on western blot (Figure 4c). There was an equal expression of EpoR and its positive control (K-562 cell lysate) while EpoR blocking peptide and Laemmli buffer in the absence of cell lysate showed no expression. There was no significant difference in the expression of EpoR between myotubes and myoblasts (P>0.05).

The expression of erythropoietin receptor (EpoR) in skeletal myotubes and myoblasts. Immunofluorescence image using Texas-Red dye (Alexa Fluor 594) shows the expression of EpoR in differentiated myotubes (a). The corresponding immunoglobulin G (IgG) isotype control shows negligible staining (b). The western blot shows the comparative expression of EpoR in C2C12 myotubes and myoblasts (c); K-562 whole-cell lysate (sc-2203) was used as positive control and EpoR blocking peptide (sc-697-p) and a lane with no cell lysates as negative controls. There was no significant difference in the expression of EpoR between C2C12 myotubes and myoblasts. Data are represented by mean±s.d., n=9. One-way analysis of variance (ANOVA)/Tukey's HSD test was used.
Simulated Ischemia Leads to Morphological Changes in Myotubes and Condensation of Nuclear Chromatin
Simulated ischemia caused shrinking of myotubes after 6–8 h (Figure 5a and b). There was an apparent increase in the number of fragmented and brightly stained nuclei as shown in Figure 5c as compared with control myotubes in Figure 5d. The quantification of apoptosis is shown in Figure 5e.

Morphological changes in the myotubes and nuclei of myotubes subjected to simulated ischemia. The myotubes appear shrunken after ischemic exposure (a), as compared with normal controls (b). There was an apparent increase in the number of fragmented and brightly stained nuclei as shown in (c) as compared with control myotubes in (d). The quantification of apoptosis is shown in (e). There were significant differences in the percentage of apoptotic nuclei at 0, 4, 6, 8 and 12 h of simulated ischemia (e). Data are represented by mean±s.d., n=9. One-way analysis of variance (ANOVA)/Tukey's HSD test was used.
There was a significant time-dependent increase in the number of apoptotic cells when myotubes were exposed to simulated ischemia using stagnant culture media (P<0.01; Figure 5e). The myotubes that were maintained in optimal normoxic conditions had less than 10% apoptotic cells as compared with 88% in the myotubes that were exposed to simulated ischemia for 12 h.
There was no inter-observer variability noted between the observations for the number of apoptotic cells made by two independent observers (Pearson's product–moment correlation coefficient, r=+0.997, n=45). When the data were plotted on a scatter chart, a strong positive correlation was shown.
It was also observed that for equivalent time periods, stagnant culture media were more effective in inducing apoptosis in myotubes than fresh DMEM containing low glucose (P<0.01) or fresh glucose-free DMEM (P<0.01). The data shown here is for 8 h using gas mixture-1 (Figure 6a). Approximately 8 h of simulated ischemia using gas mixture-1 and stagnant culture media provides adequate number of apoptotic cells for assay development and assessing the effects of compounds being tested in reducing apoptosis.

The effect of different culture medium and erythropoietin (EPO) on apoptosis. Stagnant culture media caused more profound apoptosis in combination with hypoxia for 8 h than Dulbecco's modified Eagle's medium (DMEM) low glucose or DMEM glucose-free culture medium (a). Pretreatment with EPO (60 ng/ml) caused a significant reduction in the percentage of apoptotic cells in comparison to the untreated myotubes exposed to simulated ischemia for 8 h (b). Data are represented by mean±s.d., n=9. One-way analysis of variance (ANOVA)/Tukey's HSD test was used to analyze the data in a and b.
Pretreatment with 60 ng of EPO per ml of culture medium for 24 h significantly reduced the number of apoptotic nuclei in myotubes exposed to simulated ischemia (P<0.01). The data shown is for 8 h of simulated ischemia using gas mixture-1 (Figure 6b). Similar results were achieved with ischemia exposure for 6 and 12 h (data not shown). The optimum dose of EPO (60 ng/ml of culture media) was determined through a dose–response curve, with doses ranging from 1 ng/ml to 1 μg/ml of the culture medium (data not shown).
Simulated Ischemia Led to Increased Expression of Cleaved Caspase-3 Enzyme
Immunofluorescence showed increased expression of the cleaved caspase-3 in myotubes that were exposed to simulated ischemia (Figure 7a). In addition, western blot analysis of cell lysates confirmed a significant upregulation (P<0.01) in the levels of cleaved caspase-3 enzyme in ischemic myotubes (Figure 8b). Pretreatment with EPO (60 ng/ml of culture media) caused a significant decrease in the levels of cleaved caspase-3 as shown in Figure 7a and b. The data presented in Figure 7 is from 8 h of simulated ischemia exposure.

Cleaved caspase-3 assay. The cleaved caspase-3 protein was expressed in the myotubes subjected to simulated ischemia for 8 h as shown in the immunofluorescent expression of Texas-Red dye (a) and a distinct band on western blot (b). EPO pretreatment caused a significant reduction in the expression of cleaved caspase-3 in comparison to untreated myotubes subjected to simulated ischemia (b). Data are represented by mean±s.d., n=9. One-way analysis of variance (ANOVA)/Tukey's HSD test was used to analyze the data.

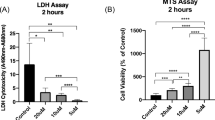
Lactate dehydrogenase (LDH) release assay. There was a significant increase in LDH release from the myotubes as measured by calorimetric assay. There were significant differences in the amount of LDH released from myotubes at 0, 4, 6, 8 and 12 h (a). Pretreatment with EPO significantly decreased the amount of LDH release from myotubes subjected to simulated ischemia for 8 h (b). Data are represented by mean±s.d., n=9. One-way analysis of variance (ANOVA)/Tukey's HSD test was used to analyze the data.
Simulated Ischemia Resulted in Increased LDH Release by Myotubes
LDH release, which is a marker of cell death, increased steadily and significantly with the duration of ischemic exposure (P<0.01). It was minimal in the culture media of myotubes maintained in optimal normoxic conditions (mean of 7.33 U/l) and maximum in those exposed to ischemia for 12 h (mean of 39.55 U/l) as shown in Figure 8a. It was also observed that EPO (60 ng/ml of culture media) significantly decreased LDH release in myotubes exposed to simulated ischemia (mean of 19.11 U/l; P<0.01). The data shown in Figure 8b is for 8 h of simulated ischemia using gas mixture-1. Significant reductions in LDH release were also noted at 4, 6 and 12 h.
DISCUSSION
This study has developed an in vitro model of myotube ischemia. It has shown that exposure of mature C2C12 myotube monolayers to nutrition depletion, hypoxia and hypercapnia for 6–12 h can simulate certain characteristics of skeletal muscle ischemia, which include a significant increase in PCO2 and decreases in PO2 and pH of the extracellular fluid. The overall effect of these changes is a significant time-dependent increase in the percentage of apoptotic cells, which is a feature of skeletal muscle ischemia.4, 5 The results also show that the most favorable duration for simulated ischemia in this model is between 6 and 12 h as it causes a relatively low pH (7.31–7.12) and high percentage of apoptotic cells (50.6–87.2%). This allows an adequate window for assay development and demonstration of the potential effects of experimental compounds.
The technique of simulated ischemia developed in this study differs from that used in previously published studies in several ways. The culture media we used to incubate the cells in the hypoxic chambers was left unchanged for 24 h (stagnant medium). This was based on the fact that the greatest depletion of nutrients including that of amino acids occurs in the first 24 h of cell culture.24, 25
In contrast to the two-dimensional model in this study, a three-dimensional model of simulated ischemia of myotubes has also been developed.26, 27 However, it may be difficult to test the tissue-protective effects of compounds in the latter model as the surface area of the myotubes exposed to the culture media and the compounds dissolved in the culture media are limited.
A gas mixture containing 80% N2 and 20% CO2 (gas mixture-1) was used to induce ischemia, instead of a gas mixture containing 95% N2, 4% CO2 and 1% O2 (gas mixture-2), which has been used in most other hypoxic models.28, 29, 30 This was based on the rationale that hypercapnia in addition to hypoxia is also an essential element of ischemia.22 It has also been shown that an increase in CO2 concentration produces a fall in the pH of culture media,31 which is essential for ischemic simulation (Table 1). As it is not possible to eliminate all of the oxygen from the system,32 gas mixture-1 (despite not having any added oxygen) still provides a hypoxic rather than an anoxic environment.
The model developed uses a hypoxic chamber (Billups-Rothenberg), which is similar to that used by other groups in previous studies.28, 29, 30 It enables a hypoxic external microenvironment to be established by altering the partial pressure of gases in the culture media. Other techniques that may be used to generate hypoxia do not offer any advantage over the hypoxic chamber when using C2C12 myotubes.33, 34, 35, 36, 37, 38
CLI is a severe form of chronic ischemia distinct from acute ischemia.1, 39 This model aims to replicate the important aspects of CLI such as apoptosis as opposed to those of acute ischemia, which consists predominantly of necrosis.40, 41 Chronic ischemia is difficult to replicate in vitro as cells will invariably undergo both apoptosis and necrosis.40 The two-dimensional model used in this study resulted in a larger proportion of cells undergoing ischemia-induced apoptosis, rather than necrosis as compared to a three-dimensional model of myotube ischemia.26, 27 This may be due to the fact that in a three-dimensional model, cells on the outside of the construct form a dense layer, which acts as a barrier to diffusion.40 As a result, adequate oxygen and nutrients are not delivered to the center of the construct where cells become more susceptible to necrosis. As the emphasis of this study was the demonstration of apoptosis and its prevention, the proportion of apoptosis and necrosis was not specifically studied.
In addition to the treatment of anemia, other non-erythropoietic roles of EPO have come to light following discovery of the expression of EpoR in extra-hemopoietic tissues.42, 43 It has also been shown that a distinct tissue protective hetero-receptor complex mediates the tissue-protective function of EPO.44 We have recently reported the expression of the tissue-protective receptor complex of EPO in human skeletal muscle.21 On the basis of this finding, the in vitro model of myotube ischemia was used to show the ability of rhEPO to decrease ischemia-induced apoptosis.
This is the first study showing the expression of EpoR in differentiated C2C12 myotubes. However, Figure 4a shows that some of the myotubes, especially those at the periphery, do not appear to express EpoR. These may be myotubes, which were non-viable at the time of fixation or may represent the limitation of the detecting system or indeed of the model itself. Ogilvie et al45 had previously shown that EpoR is expressed by C2C12 myoblasts. They also showed that EPO stimulates their proliferation and suppresses differentiation into myotubes. The results from this study indicate that EPO also protects differentiated myotubes from ischemic insult.
Lately, it has also become apparent that the use of EPO in clinical settings may be limited by thrombogenic side effects.20 However, non-erythropoietic derivatives of EPO including carbamylated EPO have now been developed and tested in pre-clinical studies.46, 47, 48, 49 It may be possible to assess these EPO derivatives in this model and investigate their potential tissue-protective properties.
In conclusion, a model system for simulated ischemia of myotubes is proposed using monolayers of C2C12 myotubes. The system is easy to use and considers important parameters of chronic ischemia, including apoptosis, PO2, PCO2 and pH. It has also been used to show that EPO decreases ischemia-induced apoptosis in C2C12 myotubes. Additional therapeutic agents designed for tissue protection in skeletal muscle ischemia can also be evaluated using this model.
References
Second European Consensus Document on chronic critical leg ischemia. Circulation 1991;84:IV1–I26.
Norgren L, Hiatt WR, Dormandy JA, et al. Inter-society consensus for the management of peripheral arterial disease (TASC II). J Vasc Surg 2007;45 (Suppl S):S5–S67.
Brass EP, Anthony R, Dormandy J, et al. Parenteral therapy with lipo-ecraprost, a lipid-based formulation of a PGE1 analog, does not alter six-month outcomes in patients with critical leg ischemia. J Vasc Surg 2006;43:752–759.
Mitchell RG, Duscha BD, Robbins JL, et al. Increased levels of apoptosis in gastrocnemius skeletal muscle in patients with peripheral arterial disease. Vasc Med 2007;12:285–290.
Aravindan N, Aravindan S, Shanmugasundaram K, et al. Periods of systemic partial hypoxia induces apoptosis and inflammation in rat skeletal muscle. Mol Cell Biochem 2007;302:51–58.
Musters RJ, Otten E, Biegelmann E, et al. Loss of asymmetric distribution of sarcolemmal phosphatidylethanolamine during simulated ischemia in the isolated neonatal rat cardiomyocyte. Circ Res 1993;73:514–523.
McPherson BC, Zhu X, Liu H, et al. Acetylcholine attenuates cardiomyocyte oxidant stress during simulated ischemia and reoxygenation. Pharmacology 2002;64:49–56.
Yuan HB, Huang Y, Zheng S, et al. Hypothermic preconditioning increases survival of Purkinje neurons in rat cerebellar slices after an in vitro simulated ischemia. Anesthesiology 2004;100:331–337.
Sochocka E, Juurlink BH, Code WE, et al. Cell death in primary cultures of mouse neurons and astrocytes during exposure to and ‘recovery’ from hypoxia, substrate deprivation and simulated ischemia. Brain Res 1994;638:21–28.
Pipinos II, Judge AR, Selsby JT, et al. The myopathy of peripheral arterial occlusive disease: part 1. Functional and histomorphological changes and evidence for mitochondrial dysfunction. Vasc Endovasc Surg 2007;41:481–489.
Pipinos II, Judge AR, Selsby JT, et al. The myopathy of peripheral arterial occlusive disease: part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc Endovasc Surg 2008;42:101–112.
Yaffe D, Saxel O . Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977;270:725–727.
Loos B, Smith R, Engelbrecht AM . Ischaemic preconditioning and TNF-alpha-mediated preconditioning is associated with a differential cPLA2 translocation pattern in early ischaemia. Prostaglandins Leukot Essent Fatty Acids 2008;78:403–413.
Lacerda L, Smith RM, Opie L, et al. TNFalpha-induced cytoprotection requires the production of free radicals within mitochondria in C2C12 myotubes. Life Sci 2006;79:2194–2201.
Dehne N, Kerkweg U, Otto T, et al. The HIF-1 response to simulated ischemia in mouse skeletal muscle cells neither enhances glycolysis nor prevents myotube cell death. Am J Physiol Regul Integr Comp Physiol 2007;293:R1693–R1701.
Ahmad R, Hand M . Recombinant erythropoietin for the anemia of chronic renal failure. N Engl J Med 1987;317:169–170.
Belonje AM, Voors AA, van Gilst WH, et al. Effects of erythropoietin after an acute myocardial infarction: rationale and study design of a prospective, randomized, clinical trial (HEBE III). Am Heart J 2008;155:817–822.
Binbrek AS, Rao NS, Al Khaja N, et al. Erythropoietin to augment myocardial salvage induced by coronary thrombolysis in patients with ST segment elevation acute myocardial infarction. Am J Cardiol 2009;104:1035–1040.
Sharples EJ, Patel N, Brown P, et al. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol 2004;15:2115–2124.
Ehrenreich H, Weissenborn K, Prange H, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke 2009;40:e647–e656.
Joshi D, Tsui J, Ho TK, et al. Review of the role of erythropoietin in critical leg ischemia. Angiology 2010;16:541–550.
Russ AL, Haberstroh KM, Rundell AE . Experimental strategies to improve in vitro models of renal ischemia. Exp Mol Pathol 2007;83:143–159.
Melo F, Carey DJ, Brandan E . Extracellular matrix is required for skeletal muscle differentiation but not myogenin expression. J Cell Biochem 1996;62:227–239.
Griffiths JB . The effect of medium changes on the growth and metabolism of the human diploid cell, W1-38. J Cell Sci 1971;8:43–52.
Glacken MW, Fleischaker RJ, Sinskey AJ . Reduction of waste product excretion via nutrient control: possible strategies for maximizing product and cell yields on serum in cultures of mammalian cells. Biotechnol Bioeng 1986;28:1376–1389.
Gawlitta D, Li W, Oomens CW, et al. The relative contributions of compression and hypoxia to development of muscle tissue damage: an in vitro study. Ann Biomed Eng 2007;35:273–284.
Gawlitta D, Oomens CW, Bader DL, et al. Temporal differences in the influence of ischemic factors and deformation on the metabolism of engineered skeletal muscle. J Appl Physiol 2007;103:464–473.
Allen CB, Schneider BK, White CW . Limitations to oxygen diffusion and equilibration in in vitro cell exposure systems in hyperoxia and hypoxia. Am J Physiol Lung Cell Mol Physiol 2001;281:L1021–L1027.
Norman JT, Clark IM, Garcia PL . Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int 2000;58:2351–2366.
Miller RL, Kohan DE . Hypoxia regulates endothelin-1 production by the inner medullary collecting duct. J Lab Clin Med 1998;131:45–48.
Gramer MJ, Ogorzalek T . A semi-empirical mathematical model useful for describing the relationship between carbon dioxide, pH, lactate and base in a bicarbonate-buffered cell-culture process. Biotechnol Appl Biochem 2007;47:197–204.
Kang Y, Greaves B, Perry RR . Effect of acute and chronic intermittent hypoxia on DNA topoisomerase II alpha expression and mitomycin C-induced DNA damage and cytotoxicity in human colon cancer cells. Biochem Pharmacol 1996;52:669–676.
Doctor RB, Bacallao R, Mandel LJ . Method for recovering ATP content and mitochondrial function after chemical anoxia in renal cell cultures. Am J Physiol 1994;266:C1803–C1811.
Dagher PC . Modeling ischemia in vitro: selective depletion of adenine and guanine nucleotide pools. Am J Physiol Cell Physiol 2000;279:C1270–C1277.
Hlatky L, Alpen EL . Two-dimensional diffusion limited system for cell growth. Cell Tissue Kinet 1985;18:597–611.
Cochran DM, Fukumura D, Ancukiewicz M, et al. Evolution of oxygen and glucose concentration profiles in a tissue-mimetic culture system of embryonic stem cells. Ann Biomed Eng 2006;34:1247–1258.
Pitts KR, Toombs CF . Coverslip hypoxia: a novel method for studying cardiac myocyte hypoxia and ischemia in vitro. Am J Physiol Heart Circ Physiol 2004;287:H1801–H1812.
Ganitkevich V, Reil S, Schwethelm B, et al. Dynamic responses of single cardiomyocytes to graded ischemia studied by oxygen clamp in on-chip picochambers. Circ Res 2006;99:165–171.
McPherson GA, Wolfe JH . ABC of vascular diseases. Acute ischaemia of the leg. BMJ 1992;304:169–172.
Majno G, Joris I . Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3–15.
Raffray M, Cohen GM . Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol Ther 1997;75:153–177.
Winearls CG . Treatment of the anaemia of chronic renal failure with recombinant human erythropoietin. Drugs 1989;38:342–345.
Maiese K, Li F, Chong ZZ . New avenues of exploration for erythropoietin. JAMA 2005;293:90–95.
Brines M, Grasso G, Fiordaliso F, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci USA 2004;101:14907–14912.
Ogilvie M, Yu X, Nicolas-Metral V, et al. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J Biol Chem 2000;275:39754–39761.
Brines M, Cerami A . Erythropoietin-mediated tissue protection: reducing collateral damage from the primary injury response. J Intern Med 2008;264:405–432.
Leist M, Ghezzi P, Grasso G, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004;305:239–242.
Fiordaliso F, Chimenti S, Staszewsky L, et al. A nonerythropoietic derivative of erythropoietin protects the myocardium from ischemia–reperfusion injury. Proc Natl Acad Sci USA 2005;102:2046–2051.
Fantacci M, Bianciardi P, Caretti A, et al. Carbamylated erythropoietin ameliorates the metabolic stress induced in vivo by severe chronic hypoxia. Proc Natl Acad Sci USA 2006;103:17531–17536.
Ihnken K, Beyersdorf F, Mitrev Z, et al. Controlled reperfusion reduces reperfusion injury in skeletal-muscle after incomplete limb ischemia. Vasc Surg 1994;28:241–259.
Hagberg H . Intracellular pH during ischemia in skeletal muscle: relationship to membrane potential, extracellular pH, tissue lactic acid and ATP. Pflugers Arch 1985;404:342–347.
Cina C, Katsamouris A, Megerman J, et al. Utility of transcutaneous oxygen tension measurements in peripheral arterial occlusive disease. J Vasc Surg 1984;1:362–371.
D’Eri A, Martini R, Cordova RM, et al. Evaluation of cutaneous necrosis risk in peripheral arterial diseases. Miner Cardioangiol 2002;50:239–244.
Brow TD, Kakkar VV, Das SK . The significance of creatine kinase in cardiac patients with acute limb ischaemia. J Cardiovasc Surg (Torino) 1999;40:637–644.
Sternbergh WC, Tuttle TM, Makhoul RG, et al. Postischemic extremities exhibit immediate-release of tumor-necrosis-factor. J Vasc Surg 1994;20:474–481.
Hiatt WR . Carnitine and peripheral arterial disease. Ann N Y Acad Sci 2004;1033:92–98.
Brevetti G, Angelini C, Rosa M, et al. Muscle carnitine deficiency in patients with severe peripheral vascular disease. Circulation 1991;84:1490–1495.
Acknowledgements
We thank Dr Jill Norman, Centre for Nephrology, UCL, Royal Free campus for provision of the hypoxic chambers; Dr Cecil Thompson, Clinical Biochemistry, UCL, Royal Free campus for help with automated measurements of LDH release and Ms Korsa Khan, UCL Centre for Rheumatology and Connective Disease for help in provision of other laboratory equipment.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
A model for the study of potential therapeutic agents for critical limb ischemia is described. Monolayers of C2C12 myoblasts can be differentiated into mature myotubes and used to simulate parameters that occur in skeletal muscle ischemia. As proof-of-concept, erythropoietin exhibits tissue protective effects in the model.
Supplementary information
Rights and permissions
About this article
Cite this article
Joshi, D., Patel, H., Baker, D. et al. Development of an in vitro model of myotube ischemia. Lab Invest 91, 1241–1252 (2011). https://doi.org/10.1038/labinvest.2011.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.79
