
Abstract
Apoptosis is a driving force of diabetic end-organ damage, including diabetic nephropathy (DN). However, the mechanisms that modulate diabetes-induced cell death are not fully understood. Heat shock protein 27 (HSP27/HSPB1) is a cell stress protein that regulates apoptosis in extrarenal cells and is expressed by podocytes exposed to toxins causing nephrotic syndrome. We investigated the regulation of HSPB1 expression and its function in podocytes exposed to factors contributing to DN, such as high glucose and angiotensin (Ang) II. HSPB1 expression was assessed in renal biopsies from patients with DN, minimal change disease or focal segmental glomerulosclerosis (FSGS), in a rat model of diabetes induced by streptozotocin (STZ) and in Ang II-infused rats. The regulation of HSPB1 was studied in cultured human podocytes and the function of HSPB1 expressed in response to pathophysiologically relevant stimuli was explored by short interfering RNA knockdown. Total kidney HSPB1 mRNA and protein expression was increased in rats with STZ-induced diabetes and in rats infused with Ang II. Upregulation of HSPB1 protein was confirmed in isolated diabetic glomeruli. Immunohistochemistry showed increased glomerular expression of HSPB1 in both models and localized glomerular HSPB1 to podocytes. HSPB1 protein was increased in glomerular podocytes from patients with DN or FSGS. In cultured human podocytes HSPB1 mRNA and protein expression was upregulated by high glucose concentrations and Ang II. High glucose, but not Ang II, promoted podocyte apoptosis. HSPB1 short interfering RNA (siRNA) targeting increased apoptosis in a high-glucose milieu and sensitized to Ang II or TGFβ1-induced apoptosis by promoting caspase activation. In conclusion, both high glucose and Ang II contribute to HSPB1 upregulation. HSPB1 upregulation allows podocytes to better withstand an adverse high-glucose or Ang II-rich environment, such as can be found in DN.
Similar content being viewed by others


Main
Diabetic nephropathy (DN) is the most frequent cause of end-stage renal disease. The diabetic milieu induces apoptosis in various end-organ systems and contributes to the gradual loss of renal function and mass in DN.1, 2, 3 Studies in humans and DN animal models reveal podocyte injury, detachment, apoptosis and loss.4, 5, 6, 7, 8 Although some stimuli induce necrosis of podocytes in culture,9 there is more in vivo evidence for a role of apoptosis in podocyte loss in DN.10, 11, 12 More specifically, podocyte apoptosis coincided with the onset of albuminuria and preceded podocytopenia in different mouse models of diabetes.13 Podocyte apoptosis has also been documented early in the course of kidney disease in patients with type 2 diabetes.14 The molecular pathways contributing to podocyte injury in DN include, but are not limited to, high glucose and activation of the local renin–angiotensin system (RAS).15 Angiotensin (Ang) II is one of the major effector molecules of the RAS and is the main target of established therapy for clinical DN.15 Intraglomerular Ang II levels are increased in DN,16 podocytes express Ang II receptors17 and local glomerular RAS directly promotes filtration barrier injury.15 Ang II promoted apoptosis of the cultured rat and mouse podocytes.18, 19, 20 However, Ang II-induced apoptosis in human podocytes is not well characterized.
Cells respond to environmental stressors by rapidly synthesizing protective proteins such as heat shock proteins (HSPs).21, 22 HSPs are ubiquitous, highly evolutionary conserved intracellular proteins categorized according to their molecular weight.23, 24 HSPs have chaperone-like activity and protect proteins from damage induced by environmental factors, such as free radicals, heat, ischemia and toxins, allowing denatured proteins to adopt their native configuration. HSPB1 is a member of the small HSP family of proteins with a molecular weight of ∼27 kDa.25 HSPB1 controls a wide range of biological activities, including actin stabilization and protection from stress and apoptosis.25, 26, 27, 28, 29, 30 HSPB1 may be of particular interest in podocyte injury because its expression is induced by the podocyte toxin puromycin aminonucleoside (PAN).31 Protective cellular responses in podocytes are poorly understood. HSPB1 regulates actin microfilament dynamics in podocyte foot processes.31 In addition, enhanced podocyte HSPB1 expression/phosphorylation has been reported in experimental nephrotic syndrome induced by the podocyte toxin PAN31 suggesting a role for HSPB1 in the pathophysiological changes of the podocyte cytoskeleton during the development of proteinuria. However, there is an incomplete understanding of the relationship between podocyte HSPB1 and stressors relevant to DN, such as high glucose and Ang II.
This study was designed to assess podocyte HSPB1 expression in vivo during kidney injury induced by diabetes or following Ang II infusion, and the role of HSPB1 on apoptosis in cultured human podocytes exposed to either high glucose or Ang II. Our results indicate that HSPB1 upregulation is a compensatory protective mechanism that prevents podocyte apoptosis in the presence of Ang II or a high-glucose milieu.
MATERIALS AND METHODS
Cell Culture and Reagents
Human podocytes are a previously described cell line32 transfected with a temperature-sensitive SV40 gene construct and a gene encoding the catalytic domain of human telomerase. At a permissive temperature of 33 °C, the cells remain in an undifferentiated proliferative state, whereas raising the temperature to 37°C results in growth arrest and differentiation to the parental podocyte phenotype. Undifferentiated podocyte cultures were maintained at 33°C in RPMI 1640 medium with penicillin, streptomycin, ITS (Insulin, transferrin, selenite) and 10% FCS. Once cells had reached 70–80% confluence, they were cultured at 37°C for at least 14 days before use, when full differentiation and nephron expression had occurred.5 Experiments were performed in differentiated cells. For experiments cells were cultured in serum-free media for 24 h before addition of stimuli and throughout the experiment. For high-glucose experiments, glucose was added in the media to reach a final concentration of 700 mg/dl vs control media with 200 mg/dl glucose. The same amount of mannitol was added as an osmolarity control. Ang II (10−7 mol/l) was added each day (Bachem). Human recombinant TGF-β1 (Peprotech, London, UK) was used at a concentration of 10 ng/ml.
Animal Models
Two groups of ten 10-week-old Wistar Kyoto rats (Criffa, Barcelona, Spain) were studied. Diabetes was induced by a single intraperitoneal 50 mg/kg streptozotocin (STZ; Sigma, St Louis, MO, USA) injection.5 Control rats received the STZ vehicle (0.01 M citrate buffer pH 4.5). Rats were killed at 7 months following induction of diabetes.5 Insulin (1–4 IU s.c., Insulatard NPH, Novo Nordisk, Denmark) was administered weekly so as to prevent death, but not with the aim of totally correcting hyperglycemia. Insulin administration was initiated 7 days after STZ, having checked that glycemia was >400 mg/dl (Glucocard, Menarini, Barcelona, Spain). Systolic blood pressure was measured monthly in conscious, restrained rats by the tail-cuff sphygmomanometer (NARCO, Biosystems, CO, USA). Albuminuria was measured by ELISA (Celltrend, Luckenwalde, Germany; Supplementary Table 1).
In an independent experiment, two groups of 10-week-old Wistar Kyoto rats (Criffa) were studied. Diabetes was induced by a single intraperitoneal 50 mg/Kg STZ injection. Rats were killed at 6 weeks following induction of diabetes. At that time albuminuria was already increased (2310±400 μg/24 h vs 440±300 μg/24 h in controls, P<0.05).
Ang II 100 ng/kg/min was systemically infused by subcutaneous osmotic minipumps (Alza Corp., CA, USA) for 24 h to 2 weeks to 3-month-old male Wistar rats (n=4–8 animals per group). A control group of same age saline-infused rats was also studied (n=8 animals).
Rats were killed and kidneys perfused in situ with cold saline before removal. One kidney was fixed in buffered formalin, embedded in paraffin and used for immunohistochemistry, and the other kidney was snap-frozen in liquid nitrogen for RNA and protein studies of renal cortexes. All experimental procedures were approved by the Animal Care and Use Committee of our Institution, according to the guidelines for ethical care of the European Community.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry and immunofluorescence were carried out in paraffin-embedded rat kidney sections 5 μm thick.33 The primary antibodies were goat polyclonal anti-HSPB1 (1:60, Santa Cruz Biotechnology, CA, USA) and FITC-mouse anti-synaptopodin (1:10, Progen, Heidelberg, Germany). Secondary HRP- and phycoerythrin-conjugated antibodies were used for HSPB1. For immunohistochemistry sections were counterstained with Carazzi's hematoxylin. Negative controls included incubation with a non-specific immunoglobulin of the same isotype as the primary antibody.
Western Blot
Western blots were performed as described previously.34 Membranes were incubated overnight at 4°C with rabbit polyclonal anti-HSPB1 antibody (1:500; Santa Cruz Biotechnology), mouse monoclonal anti-HSP70 and anti-HSP47 (1:1000, SPA-810, Stressgen), rabbit polyclonal anti-cleaved caspase-3 (1:1000; Cell Signaling, Hertfordshire, UK) or mouse anti-tubulin monoclonal antibody (1:5000, Sigma) followed by incubation with horseradish peroxidase-conjugated secondary antibody (1:2000, Amersham, Aylesbury, UK). Blots were developed with the enhanced chemiluminescence method (ECL) following the manufacturer's instructions (Amersham).
Isolation of Glomerular Protein
Kidneys from the 7-month diabetic rats and their control were harvested, decapsulated and macerated through three sieves (150, 106 and 63 μm). Tissue trapped by the 63 μm sieve was washed with PBS and centrifuged at 4°C for 5 min. The pellet was resuspended in PBS and examined under phase contrast microscopy. The preparation contained >95% glomeruli with minimal tubular contamination. Then, western blot was performed as described previously.
Real-Time Reverse Transcription-PCR
RNA was isolated by Trizol (Invitrogen, Paisley, UK). A total of 1 μg of RNA was reverse transcribed with the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR reactions were performed on an ABI Prism 7500 sequence detection PCR system (Applied Biosystems) according to the manufacturer's protocol using the DeltaDelta Ct method.35 Expression levels are given as ratios to GAPDH. Pre-developed primer and probe assays (PDAR) were obtained for human GAPDH and HSPB1 from Applied Biosystems.
Confocal Microscopy
Cells plated onto Labtek slides were fixed in 4% paraformaldehyde and permeabilized in 0.2%. Triton X-100 in PBS for 10 min each. After washing in PBS cells were incubated overnight at 4°C with goat polyclonal anti-HSPB1 antibody (1:50, Santa Cruz) followed by incubation with anti-goat Alexa Fluor 488 (1:300, Invitrogen). The cell nuclei were counterstained with DAPI (Vector Laboratories, Burlingame, CA, USA) to observe the typical morphological changes. After washing, cells were mounted in 70% glycerol in PBS, and analyzed with a DM-IRB confocal microscope (Leica DM, Bannockburn, IL, USA).36
Cell Death and Apoptosis
Cells were cultured to subconfluence in 12-well plates. Apoptosis was assessed by functional and morphological studies. Flow cytometry of DNA content was used to quantitate apoptosis. Adherent cells were pooled with spontaneously detached cells, and stained in 100 μg/ml propidium iodide, 0.05% NP-40, 10 μg/ml RNAse A in PBS and incubated at 4°C for >1 h. This assay permeabilizes the cells. Permeabilization allows entry of propidium iodide in all cells, dead and alive. Apoptotic cells are characterized by a lower DNA content (hypodiploid cells) because of nuclear fragmentation. Thus, this assay is not based on the known ability of propidium iodide to enter dead cells. The percentage of apoptotic cells with decreased DNA content (A0) was counted.37, 38 Cells were exposed to high (700 mg/dl) glucose, Ang II (10–7 M) or TGFβ1 (10 ng/ml).
To assess the typical apoptotic nuclear changes cells were cultured in chamber slides (Labtek, Nunc, Naperville, IL, USA), fixed with methanol:acetone (1:1) and stained with DAPI (Sigma) and anti-HSPB1. The typical condensed, shrunk and fragmented nuclei of apoptotic cells were identified by a laser scanning confocal microscope (Leika).
The ApoScreen Annexin V-FITC kit (Southern Biotechnology Associates, Birmingham, AL, USA) was used to assess both apoptosis and necrosis by flow cytometry. This method discriminates between early apoptotic (propidium iodide negative, annexin V positive), and late apoptotic or necrotic cells (propidium iodide positive, annexin V positive).39
Caspase-9 activity was assessed in cells transfected with HSPB1 siRNA and stimulated with Ang II or high glucose. Caspase-9 activity (R&D Systems, Minneapolis, MN, USA) was measured following the manufacturer’s instructions. In brief, cell extracts (100 μg protein) were incubated in half-area 96-well plates at 37°C with 200 μ M LEDH-pNA peptide in a total volume of 100 μl. The pNA light emission was quantified using a spectrophotometer plate reader at 405 nm. Comparison of the absorbance of pNA from an apoptotic sample with an uninduced control allows determination of the fold increase in caspase activity.
Transfection of Small Interfering RNA
Cells were grown in six-well plates (Costar, Cambridge, MA, USA) and transfected with a mixture of 25 nmol/ml HSPB1 siRNA (Santa Cruz), Opti-MEM I Reduced Serum Medium and Lipofectamine 2000 (Invitrogen).5 Two independent siRNAs were used and similar results were obtained. After 18 h, cells were washed and cultured for 24 h in a complete medium, and serum depleted for 24 h before addition of high glucose or Ang II. This time point was selected from a time-course of decreasing HSPB1 protein expression in response to siRNA. A negative control scrambled siRNA provided by the manufacturer did not reduce HSPB1 protein.
Human Renal Biopsies
Kidney samples were obtained from type 2 diabetes mellitus (n=8) patients biopsied because of nephrotic proteinuria. Clinical data at biopsy were as follows: mean age 54 years (range 28–78 years); mean serum creatinine 2.5 mg/dl (range 1.1–3.6 mg/dl); mean proteinuria 6.1 g/day (range 1.0–14.5 g/day). Biopsies showed a clear picture of diffuse or nodular glomerulosclerosis with a marked tubulointerstitial involvement. Normal kidneys were also assessed. Minimal change disease (MCD) was used as a non-progressive proteinuric control (n=9: mean age 18 years (range 2–56 years); mean serum creatinine 0.78 mg/dl (range 0.34–2.5 mg/dl); mean proteinuria 4.61 g/day (range 0.3–14 g/day). Focal segmental glomerulosclerosis (FSGS) was studied as a progressive proteinuric disease (n=9: mean age 34 years (range 13–57 years); mean serum creatinine 1.0 mg/dl (range 0.51–1.7 mg/dl); mean proteinuria 5.2 g/day (range 0.75–9.6 g/day). The study protocol was approved by the local Ethics Committee and an informed consent was obtained.
Immunohistochemistry was carried out in paraffin-embedded human kidney, fixed in 4% buffered formalin or Bouin. The primary antibody was goat polyclonal anti-HSPB1 (1:5000, Santa Cruz biotechnology). Briefly, 5-μm-thick renal sections were adhered to polylysine-coated glass slides and fixed overnight at 56°C. After deparaffinizing through xylene, alcohol and distilled water, endogenous peroxidase was blocked by 3% H2O2 for 10 min. Sections were microwaved in 0.1 mM citrate buffer pH 6.0 for 20 min, and transferred to distilled water. After rinsing in Tris-HCl–phosphate-buffered saline (TPS), sections were incubated with 1:10 normal rabbit serum in TPS/1% BSA and then incubated overnight at 4°C with unlabeled goat anti-human HSPB1. Next, they were incubated with biotinylated rabbit anti-goat antibody (1:500) in TPS–1% BSA for 30 min at 22°C. After three rinses in TPS, they were incubated with streptavidin–peroxidase (Dako) 1:500 for 30 min. Color was developed with diaminobenzidine and then counterstained with haematoxylin, dehydrated and mounted with the Canadian balsam (Polysciences). Specificity was checked by omission of primary antibodies and use of non-immune sera.
Statistics
Data are given as mean±s.d. The Mann–Whitney, two-sided t-test or one-way ANOVA were applied to indicate significantly different mean values in comparison with the control group. A P-value < 0.05 was considered statistically significant.
RESULTS
Increased HSPB1 Expression in Experimental Diabetic Nephropathy
Increased podocyte HSPB1 had been observed in rats with nephrotic syndrome induced by PAN.25 However, this toxic model has no obvious human counterpart and it was unknown whether similar changes occur in subnephrotic proteinuria or podocyte injury induced by endogenous pathophysiological stimuli. We studied a chronic rat model of DN induced by a single STZ injection and characterized by the development of albuminuria (1071±247 vs 390±70 μg/24 h, P<0.02, at 7 months, Supplementary Table 1). At this time point whole kidney HSPB1 mRNA (Figure 1a) and protein (Figure 1b) levels were increased. The glomerular HSPB1 mRNA expression was significantly increased in DN compared with control rats (Figure 1c). Immunohistochemistry also localized increased HSPB1 expression to glomeruli of DN rats (Figure 1d), where HSPB1 co-localized with anti-synaptopodin-positive podocytes (Figure 1e). In an early rat model of DN (6 weeks) in which albuminuria was starting to increase, immunohistochemistry showed an increased glomerular HSPB1 expression (Figure 2a).

Expression of HSPB1 in experimental diabetic nephropathy. (a) Whole kidney HSPB1 mRNA was increased in diabetic rats when compared with controls. Mean±s.d. of 10 rats per group. *P<0.04 vs control rats. (b) Quantification and representative image of western blot of whole kidney protein. Western blot quantification expressed as percentage increase over control. Mean±s.d. of 10 rats per group. *P<0.006 vs control rats. (c) Western blot analysis of glomerular HSPB1. Mean±s.d. of 10 rats per group. *P<0.03 vs control rats. (d) Immunohistochemistry, representative images of control and DN kidney in which podocyte staining for HSPB1 is observed (arrows). Original magnification × 200 and × 400. (e) Double anti-synaptopodin (green) and anti-HSPB1 (red) staining of glomeruli in rat DN. Anti-synaptopodin stains mainly the foot processes and less intensely the podocyte body and the area near the cell membrane, whereas anti-HSPB1 stains mainly the podocyte body. Several clearly identifiable podocytes are indicated by arrows. Original magnification × 400.


Renal HSPB1 expression is upregulated in early diabetic nephropathy and chronic Ang II infusion. (a) HSPB1 immunohistochemistry, representative images of control and DN rat kidney at week 6 following injection of streptozotocin or vehicle. Podocyte staining for HSPB1 is observed in DN. Original magnification × 200 and × 400. (b) Whole kidney HSPB1 mRNA was increased in Ang II-infused rats when compared with controls. Mean±s.d. of 6–8 animals per group. *P<0.008 vs control. (c) Quantification and representative image of western blot of whole kidney protein. Mean±s.d. of 6–8 animals per group. *P<0.01 vs control. (d) Representative immunohistochemistry where HSPB1 is increased in the glomeruli in a distribution suggestive of podocyte expression. Original magnification × 200 and × 400.
Chronic Ang II Infusion in Rats Increased Kidney HSPB1 Expression
Ang II is a key determinant of DN. Ang II infusion in rodents causes renal damage, characterized by glomerular and interstitial inflammatory cell infiltration, observed at 3 days, and tubular atrophy and interstitial fibrosis found after 2 weeks.40, 41, 42 At this time point, Ang II increased total kidney HSPB1mRNA and protein levels, as assessed by real-time PCR (Figure 2b) and western blot (Figure 2c), respectively. Immunohistochemistry localized HSPB1 expression to glomeruli, mainly in podocytes (Figure 2d).
Increased Expression of HSPB1 in Human Diabetic Nephropathy
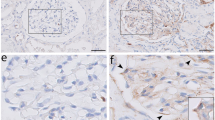
HSPB1 was studied in normal human kidneys, MCD, DN and FSGS. MCD is a non-progressive proteinuric kidney diseases, whereas both DN and FSGS are characterized by progressive glomerular injury and podocyte loss. HSPB1 expression was absent from normal glomeruli or MCD podocytes (Figures 3a and b). Increased glomerular HSPB1 expression was noted by immunohistochemistry in all human DN and FSGS biopsies and morphologically localized to podocytes (Figures 3c and d). HSPB1 tubular staining was observed in FSGS and DN and in occasional tubules in MCD.

Increased HSPB1 expression in human diabetic nephropathy biopsies. (a) Representative images of normal kidneys. (b) Minimal change disease (MCD). (c) Diabetic nephropathy (DN). (d) Focal segmental glomerulosclerosis (FSGS). HSPB1 immunohistochemistry. Note a marked increase in HSPB1 staining in glomerular cells with a podocyte localization (arrow) in DN and FSGS. Original magnification × 200 and × 400.
Regulation of HSPB1 in Cultured Human Podocytes
HSPB1 is upregulated in response to stress.43 Because HSPB1 was upregulated in human DN we tested the hypothesis that stressed human podocytes could synthesize HSPB1 as a defense mechanism. Based on experimental model results, we tested the regulation of HSPB1 expression by high glucose or Ang II, two of the main contributors to the pathogenesis of human DN. Either a high glucose concentration or Ang II increased HSPB1 mRNA (Figures 4a and c) and protein expression in human podocytes (Figures 4b and d). Mannitol, used as an osmolarity control, did not change HSPB1 expression (Figure 4a).

High glucose and Ang II increase HSPB1 expression in cultured human podocytes. Cells were exposed to (a, b) high glucose or (c, d) Ang II. High glucose (a) or Ang II (c) increases HSPB1 mRNA in a time-dependent manner. Real-time RT-PCR. Mean±s.d. of three independent experiments. *P<0.03 vs control 24 h. (b, d) Quantification and representative western blot of HSPB1. Cells were cultured in the presence of (b) high glucose and (d) Ang II for 24 h. Mean±s.d. of three independent experiments. *P<0.04 vs control.
HSPB1 Expression is a Compensatory Mechanism That Protects Human Podocytes From High Glucose- or Ang II-Induced Apoptosis
Once demonstrated that HSPB1 expression is upregulated by high glucose and Ang II in vivo and in cell culture, we approached the function of HSPB1 under these conditions. That is, we assessed the function of the increased HSPB1 levels observed in response to an environment found in DN in vivo. High glucose induces human podocyte stress and apoptosis.5 To investigate the function of HSPB1 in human podocytes exposed to high glucose, HSPB1 was knocked down by using specific siRNAs. Two different siRNAs (siRNA1 and siRNA2) were used. Results using siRNA1 are shown in Figures 5b, d, e, and 6b, c. Results using siRNA2 are shown in Figures 5a, c and 6a. Using siRNA1 we obtained similar results to those with siRNA2 shown in Figures 5a, c and 6a (not shown). Western blot confirmed efficient gene silencing of HSPB1 in the absence of changes in the levels of other HSP proteins (Figure 5a). HSPB1 targeting did not modulate cell death in non-stressed cells (Figure 5b). However, HSPB1 knockdown increased the apoptosis rate, assessed as hypodiploid cells, of podocytes stressed by a high-glucose environment (Figure 5b). Ang II had a negligible effect on human podocyte apoptosis. However, preventing HSPB1 upregulation sensitized to Ang II-induced apoptosis (Figure 5b). Western blot and confocal microscopy confirmed HSPB1 upregulation by high glucose and Ang II at the individual cell level, as well as effective HSPB1 downregulation by specific siRNAs (Figure 5c and d). Assessment and quantification of morphological features of apoptosis confirmed the apoptosis sensitizing effect of HSPB1 targeting in the presence of high glucose or Ang II (Figure 5e). The results were confirmed using a different siRNA and method to assess apoptosis. In this regard, the percentage of apoptotic cells assessed as propidium iodide negative, annexin V-positive cells was higher after HSPB1 knockdown with HSPB1 siRNA (Figure 6a). HSPB1 targeting also increased TGFβ1-induced apoptosis (Figure 6a). These results suggest that HSPB1 upregulation in the presence of DN environmental stressors protects human podocytes from stress induced by stimuli present in the DN environment.

Inhibition of HSPB1 expression by siRNA sensitizes to apoptosis in human podocytes. (a) HSPB1 siRNA downregulation of HSPB1. Representative western blot of the specific siRNA. (b) HSPB1 knockdown with HSPB1 siRNA increases high glucose-induced apoptosis and promotes Ang II-induced apoptosis. Cell death was assessed by flow cytometry of DNA content (hypodiploid cells) after culture in the presence of high glucose or Ang II (10−7 M) for 24 h. Mean±s.d. of three independent experiments. *P<0.05 vs control; **P<0.001 vs high glucose; #P<0.03 vs Ang II alone. (c) Representative western blot corresponding to the conditions of the experiment shown in b. (d) HSPB1 silencing increased apoptosis in human podocytes as assessed by morphology. Representative image of apoptotic cells with typical nuclei (shrunk, bright; DAPI staining) among cells transfected with HSPB1 siRNA (arrows). Anti-HSPB1 staining in green confirmed upregulation of HSPB1 expression by high glucose or Ang II and effective downregulation by siRNA. (e) Quantification of apoptotic cells expressed as percentage of cells with apoptotic nuclei as assessed by DAPI staining. Mean±s.d. for three independent experiments. *P<0.009 vs high glucose and Ang II alone; **P<0.0001 vs control; #P<0.04 vs control.

Inhibition of HSPB1 expression by siRNA sensitizes to apoptosis promoting caspase activation in human podocytes. (a) HSPB1 knockdown with HSPB1 siRNA increases high glucose- and TGFβ1-induced apoptosis and promotes Ang II-induced apoptosis. Cell death was assessed by flow cytometry following staining with propidium iodide and annexin V after culture in the presence of high glucose, Ang II (10−7 M) or TGFβ1 (10 ng/ml) for 24 h. Representative experiment. (b) Podocyte apoptosis induced by HSPB1 knocked down with HSPB1 siRNA is associated with caspase activation. Caspase-3 is processed in the course of Ang II- and high glucose-induced apoptosis when HSPB1 is knocked down. Quantification of active caspase 3 western blot. Mean±s.d. for three independent experiments *P<0.001 vs angiotensin, **P<0.004 vs high glucose. (c) HSPB1 knockdown promotes caspase-9 activity in human podocytes exposed to Ang II. The mean±s.d. for three independent experiments *P<0.02 vs control, **P<0.05 vs angiotensin alone.
HSPB1 Targeting Promotes Caspase Activation in Podocytes
In the mitochondrial pathway for apoptosis release of cytochrome c from the mitochondria promotes activation of caspase-9.44 Caspase-9 promotes activation of downstream caspases, such as caspase-3, the central executioner caspase, common to several apoptotic pathways. HSPB1 targeting using specific siRNAs promoted caspase-3 activation in podocytes stressed by Ang II or by a high-glucose environment (Figure 6b). By contrast, caspase-9 activity was increased only in podocytes stressed by Ang II (Figure 6c) but not in those stressed by a high-glucose environment, suggesting that these stressors activate different apoptotic pathways that have in common the regulation by HSPB1.
DISCUSSION
The role of HSPB1 on podocytes exposed to stressors relevant for DN has not been well characterized. We now show that in podocytes HSPB1 is upregulated in vivo and in culture by stimuli relevant for the pathogenesis of DN, ie high glucose and Ang II. Indeed, podocyte HSPB1 is increased in human DN. HSPB1 upregulation appears to be a compensatory mechanism that prevents or reduces podocyte apoptosis induced by high glucose or Ang II. In this regard, HSPB1 targeting, effectively preventing HSPB1 upregulation by these stimuli, sensitized to apoptosis podocytes stressed by stimuli relevant to DN such as high glucose, Ang II and TGFβ1.
HSPB1 is not a classic chaperone as it does not actively refold other proteins. It instead has a chaperone-like activity, preventing aggregation of other partially denatured proteins. Thus, HSPB1 interacts with a large number of different proteins and prevents cell death caused by hyperthermia, oxidative stress, staurosporine, Fas ligand and cytotoxic drugs.24, 28, 45, 46, 47, 48, 49 HSPB1 is an antioxidant and inhibits multiple steps in the extrinsic and intrinsic mitochondrial apoptotic pathways. However, HSPB1 target molecules differ for different lethal stimuli and cell types.50, 51 In certain cells HSPB1 binds to cytochrome c released from the mitochondria and HSPB1 overerexpression prevents cytochrome-c-mediated interaction of Apaf-1 with procaspase-9 and procaspase-9 activation.28, 30 In this regard, in human podocytes we observed that HSPB1 targeting promoted caspase-9 and caspase-3 activity in the presence of Ang II. Caspase-3 may be a downstream target of caspase-9 or other activator caspases.44 However, despite no change in caspase-9 activity, HSPB1 targeting increased caspase-3 activation in podocytes exposed to high glucose. This suggests caspase-9-independent effects of HSPB1. In this sense, in oocytes HSPB1 targeting promoted caspase-3 activation in the absence of caspase-9 activation, similar to the observation in podocytes exposed to high-glucose conditions.52
Podocyte injury results in proteinuric renal disease.53 Contrary to MCD, both DN and FSGS are progressive glomerular diseases characterized by progressive loss of podocytes in which podocyte apoptosis has been observed.13, 14 Podocyte HSP27 expression was found in DN or FSGS, but not in normal kidney or MCD. This suggests that DN and FSGS may share podocyte stressors that result in podocyte loss and that engage podocyte adaptive responses and that these adaptive responses are not engaged in MCD. The precise factors contributing to podocyte HSPB1 expression in FSGS may include Ang II itself, because overexpression of the AT1 receptor in podocytes results in FSGS.54 It is interesting to note that in experimental nephrotic syndrome caused by toxin-mediated podocyte injury, HSP25 (the mouse homolog of human HSPB1) levels increased,31 and enforced overexpression of HSP25 in cultured murine podocytes protected the actin cytoskeleton against toxin-induced disruption.55 Previous studies had observed a transient increase in glomerular HSPB1 phosphorylation in short-term diabetes and p38MAPK-dependent HSPB1 phosphorylation in murine podocytes exposed to high glucose, but the role of HSPB1 in glucose-induced apoptosis was not studied.55 HSPB1 may become phosphorylated at different serines in response to heat shock and various stimuli (reviewed in Kostenko and Moens56). The p38 MAPK pathway has a key role in HSPB1 phosphorylation, also in podocytes.57 Indeed, high glucose promotes HSPB1 phosphorylation in renal cells in an MK(MAPKAP)-2-dependent manner.58 The phosphorylation state of HSPB1 can influence its function. Thus, it modulates the size of HSPB1 oligomers and facilitates the nuclear migration of the protein.59 HSPB1 phosphorylation may influence the fate of the cell in response to an apoptotic stimulus, although the requirement for phosphorylation may vary in different systems.60 A phosphorylation-defective mutant HSPB1 accelerated toxin-induced apoptosis in renal epithelial cells.61 Overexpression of the wild-type HSPB1, but not a non-phosphorylatable mutant, markedly reduced complement-mediated glomerular epithelial cell injury.62 Indeed, increased glomerular HSPB1 phosphorylation was noted in experimental toxin-induced nephrotic syndrome and diabetes, but decreased in ischemia.58, 63, 64 However, prevention of renal HSPB1 phosphorylation did not interfere with the development of early DN in diabetic mice.58
In renal tubular cells, HSPB1 overexpression inhibited apoptosis and HSPB1 knockdown promoted apoptosis in response to culture in glucose-free medium containing sodium cyanide and 2-deoxy-D-glucose.51 However, this kind of metabolic stress is of dubious clinical significance in the particular setting of DN. We now show that glomerular HSPB1 is upregulated in humans and in rats with short- (6 weeks) and long-standing (7 months) DN and that podocytes are the key sources of glomerular HSPB1 in both clinical and experimental DN. This appears to be the consequence, at least in part, of high glucose levels, because high glucose upregulated HSPB1 in cultured human podocytes. High glucose is known to promote renal cell apoptosis, including podocyte apoptosis.3, 5 We have observed that impairing the HSPB1 upregulation adaptive response by siRNA targeting sensitizes to high glucose-induced apoptosis, while having a negligible effect on spontaneous apoptosis. Thus, these studies expand previous data by showing that stimuli pathophysiologically relevant to DN, such as high glucose, increase podocyte HSPB1 and also by showing that prevention of this upregulation promotes apoptosis. Thus, even when not reaching very high HSPB1 levels as a consequence of enforced expression in transiently transfected cells, HSPB1 is also protective.
In DN high levels of glucose recruit secondary mediators of injury that aggravate and perpetuate renal injury. Ang II is the secondary mediator of DN with most clinical relevance. Ang II has hormonal as well as paracrine/autocrine functions that modulate renal injury. Overexpression of podocyte Ang II type 1 receptor in rats results in proteinuria, nephron loss and glomerulosclerosis.54 Inhibition of RAS activity lowers proteinuria and slows progression to end-stage renal disease, and is the keystone of human DN therapy.65, 66 HSPB1 had previously been shown to be upregulated in the kidney by short-term (<1 week) Ang II infusion.67 However, the glomerular location of HSPB1 upregulation was not described and the function of HSPB1 was not addressed.67 We now show glomerular HSPB1 upregulation in a model of chronic infusion of Ang II, in vivo localization of HSPB1 expression to podocytes, and a direct effect of Ang II on cultured podocytes promoting HSPB1 upregulation. As in the case of high glucose exposure, inhibition of HSPB1 expression promotes Ang II-induced apoptosis. Thus, upregulation of HSPB1 in response to Ang II is a podocyte adaptive response to prevent Ang II- and also TGFβ1-induced cell death. Further studies should explore whether HSPB1 might also regulate additional proinflammatory actions of Ang II.68
In conclusion, HSPB1 is upregulated in vivo and in cultured podocytes by high glucose and Ang II. This appears to be an adaptive response that allows podocytes to cope with these stressors and prevents apoptosis because depletion of this upregulated HSPB1 sensitized to these same stimuli. Increased amounts of endogenous HSPB1 are required to prevent podocyte apoptosis in a diabetic milieu rich in glucose and Ang II. This may lead to the development of new therapeutic approaches.
References
Adeghate E . Molecular and cellular basis of the aetiology and management of diabetic cardiomyopathy: a short review. Mol Cell Biochem 2004;261:187–191.
Kowluru RA . Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal 2005;7:1581–1587.
Ortiz A, Ziyadeh FN, Neilson EG . Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J Investig Med 1997;45:50–56.
Jefferson JA, Shankland SJ, Pichler RH . Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int 2008;74:22–36.
Sanchez-Nino MD, Sanz AB, Ihalmo P, et al. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol 2009;20:353–362.
Reddy GR, Kotlyarevska K, Ransom RF, et al. The podocyte and diabetes mellitus: is the podocyte the key to the origins of diabetic nephropathy? Curr Opin Nephrol Hypertens 2008;17:32–36.
Wolf G, Chen S, Ziyadeh FN . From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 2005;54:1626–1634.
Steffes MW, Schmidt D, McCrery R, et al. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int 2001;59:2104–2113.
Sieber J, Lindenmeyer MT, Kampe K, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol 2010;299:F821–F829.
Sanchez-Nino MD, Benito-Martin A, Ortiz A . New paradigms in cell death in human diabetic nephropathy. Kidney Int 2010;78:737–744.
Spurney RF, Coffman TM . Stressed-out podocytes in diabetes? J Am Soc Nephrol 2008;19:2035–2037.
Niranjan T, Bielesz B, Gruenwald A, et al. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med 2008;14:290–298.
Susztak K, Raff AC, Schiffer M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006;55:225–233.
Verzola D, Gandolfo MT, Ferrario F, et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int 2007;72:1262–1272.
Xu ZG, Miao LN, Cui YC, et al. Angiotensin II type 1 receptor expression is increased via 12-lipoxygenase in high glucose-stimulated glomerular cells and type 2 diabetic glomeruli. Nephrol Dial Transplant 2009;24:1744–1752.
Ruster C, Bondeva T, Franke S, et al. Angiotensin II upregulates RAGE expression on podocytes: role of AT2 receptors. Am J Nephrol 2009;29:538–550.
Sharma M, Sharma R, Greene AS, et al. Documentation of angiotensin II receptors in glomerular epithelial cells. Am J Physiol 1998;274:F623–F627.
Ding G, Reddy K, Kapasi AA, et al. Angiotensin II induces apoptosis in rat glomerular epithelial cells. Am J Physiol Renal Physiol 2002;283:F173–F180.
Jia J, Ding G, Zhu J, et al. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol 2008;28:500–507.
Zhang H, Ding J, Fan Q, et al. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med (Maywood) 2009;234:1029–1036.
Aufricht C, Ardito T, Thulin G, et al. Heat-shock protein 25 induction and redistribution during actin reorganization after renal ischemia. Am J Physiol 1998;274:F215–F222.
Kim HP, Morse D, Choi AM . Heat-shock proteins: new keys to the development of cytoprotective therapies. Expert Opin Ther Targets 2006;10:759–769.
Fink AL . Chaperone-mediated protein folding. Physiol Rev 1999;79:425–449.
Arrigo AP . sHsp as novel regulators of programmed cell death and tumorigenicity. Pathol Biol (Paris) 2000;48:280–288.
Ferns G, Shams S, Shafi S . Heat shock protein 27: its potential role in vascular disease. Int J Exp Pathol 2006;87:253–274.
Lavoie JN, Lambert H, Hickey E, et al. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol 1995;15:505–516.
Vertii A, Hakim C, Kotlyarov A, et al. Analysis of properties of small heat shock protein Hsp25 in MAPK-activated protein kinase 2 (MK2)-deficient cells: MK2-dependent insolubilization of Hsp25 oligomers correlates with susceptibility to stress. J Biol Chem 2006;281:26966–26975.
Garrido C, Bruey JM, Fromentin A, et al. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J 1999;13:2061–2070.
Jaattela M . Overexpression of major heat shock protein hsp70 inhibits tumor necrosis factor-induced activation of phospholipase A2. J Immunol 1993;151:4286–4294.
Bruey JM, Ducasse C, Bonniaud P, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol 2000;2:645–652.
Smoyer WE, Gupta A, Mundel P, et al. Altered expression of glomerular heat shock protein 27 in experimental nephrotic syndrome. J Clin Invest 1996;97:2697–2704.
Saleem MA, O′Hare MJ, Reiser J, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 2002;13:630–638.
Sanchez-Nino MD, Sanz AB, Lorz C, et al. BASP1 promotes apoptosis in diabetic nephropathy. J Am Soc Nephrol 2010;21:610–621.
Lorz C, Benito-Martin A, Boucherot A, et al. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol 2008;19:904–914.
Sanz AB, Justo P, Sanchez-Nino MD, et al. The cytokine TWEAK modulates renal tubulointerstitial inflammation. J Am Soc Nephrol 2008;19:695–703.
Justo P, Sanz AB, Egido J, et al. 3,4-Dideoxyglucosone-3-ene induces apoptosis in renal tubular epithelial cells. Diabetes 2005;54:2424–2429.
Catalan MP, Reyero A, Egido J, et al. Acceleration of neutrophil apoptosis by glucose-containing peritoneal dialysis solutions: role of caspases. J Am Soc Nephrol 2001;12:2442–2449.
Catalan MP, Santamaria B, Reyero A, et al. 3,4-di-deoxyglucosone-3-ene promotes leukocyte apoptosis. Kidney Int 2005;68:1303–1311.
Catalan MP, Esteban J, Subira D, et al. Inhibition of caspases improves bacterial clearance in experimental peritonitis. Perit Dial Int 2003;23:123–126.
Carvajal G, Rodriguez-Vita J, Rodrigues-Diez R, et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int 2008;74:585–595.
Ruiz-Ortega M, Lorenzo O, Ruperez M, et al. Systemic infusion of angiotensin II into normal rats activates nuclear factor-kappaB and AP-1 in the kidney: role of AT(1) and AT(2) receptors. Am J Pathol 2001;158:1743–1756.
Ruperez M, Ruiz-Ortega M, Esteban V, et al. Angiotensin II increases connective tissue growth factor in the kidney. Am J Pathol 2003;163:1937–1947.
Snoeckx LH, Cornelussen RN, Van Nieuwenhoven FA, et al. Heat shock proteins and cardiovascular pathophysiology. Physiol Rev 2001;81:1461–1497.
Sanz AB, Santamaria B, Ruiz-Ortega M, et al. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol 2008;19:1634–1642.
Mehlen P, Briolay J, Smith L, et al. Analysis of the resistance to heat and hydrogen peroxide stresses in COS cells transiently expressing wild type or deletion mutants of the Drosophila 27-kDa heat-shock protein. Eur J Biochem 1993;215:277–284.
Mehlen P, Schulze-Osthoff K, Arrigo AP . Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death. J Biol Chem 1996;271:16510–16514.
Oesterreich S, Weng CN, Qiu M, et al. The small heat shock protein hsp27 is correlated with growth and drug resistance in human breast cancer cell lines. Cancer Res 1993;53:4443–4448.
Bruey JM, Paul C, Fromentin A, et al. Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 2000;19:4855–4863.
Charette SJ, Landry J . The interaction of HSP27 with Daxx identifies a potential regulatory role of HSP27 in Fas-induced apoptosis. Ann N Y Acad Sci 2000;926:126–131.
Paul C, Simon S, Gibert B, et al. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp Cell Res 2010;316:1535–1552.
Havasi A, Li Z, Wang Z, et al. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem 2008;283:12305–12313.
Liu JJ, Ma X, Cai LB, et al. Downregulation of both gene expression and activity of Hsp27 improved maturation of mouse oocyte in vitro. Reprod Biol Endocrinol 2010;8:47.
Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 2005;16:2941–2952.
Hoffmann S, Podlich D, Hahnel B, et al. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol 2004;15:1475–1487.
Dai T, Natarajan R, Nast CC, et al. Glucose and diabetes: effects on podocyte and glomerular p38MAPK, heat shock protein 25, and actin cytoskeleton. Kidney Int 2006;69:806–814.
Kostenko S, Moens U . Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci 2009;66:3289–3307.
Barutta F, Pinach S, Giunti S, et al. Heat shock protein expression in diabetic nephropathy. Am J Physiol Renal Physiol 2008;295:F1817–F1824.
Park JK, Ronkina N, Höft A, et al. Deletion of MK2 signalling in vivo inhibits small Hsp phosphorylation but not diabetic nephropathy. Nephrol Dial Transplant 2008;23:1844–1853.
Bryantsev AL, Chechenova MB, Shelden EA . Recruitment of phosphorylated small heat shock protein Hsp27 to nuclear speckles without stress. Exp Cell Res 2007;313:195–209.
Hollander JM, Martin JL, Belke DD, et al. Overexpression of wild-type heat shock protein 27 and a nonphosphorylatable heat shock protein 27 mutant protects against ischemia/reperfusion injury in a transgenic mouse model. Circulation 2004;110:3544–3552.
de Graauw M, Tijdens I, Cramer R, et al. Heat shock protein 27 is the major differentially phosphorylated protein involved in renal epithelial cellular stress response and controls focal adhesion organization and apoptosis. J Biol Chem 2005;280:29885–29898.
Aoudjit L, Stanciu M, Li H, et al. p38 mitogen-activated protein kinase protects glomerular epithelial cells from complement-mediated cell injury. Am J Physiol Renal Physiol 2003;285:F765–F774.
Smoyer WE, Ransom R, Harris RC, et al. Ischemic acute renal failure induces differential expression of small heat shock proteins. J Am Soc Nephrol 2000;11:211–221.
Smoyer WE, Ransom RF . Hsp27 regulates podocyte cytoskeletal changes in an in vitro model of podocyte process retraction. FASEB J 2002;16:315–326.
Benigni A, Gagliardini E, Remuzzi G . Changes in glomerular perm-selectivity induced by angiotensin II imply podocyte dysfunction and slit diaphragm protein rearrangement. Semin Nephrol 2004;24:131–140.
Macconi D, Bonomelli M, Benigni A, et al. Pathophysiologic implications of reduced podocyte number in a rat model of progressive glomerular injury. Am J Pathol 2006;168:42–54.
Ishizaka N, Aizawa T, Ohno M, et al. Regulation and localization of HSP70 and HSP25 in the kidney of rats undergoing long-term administration of angiotensin II. Hypertension 2002;39:122–128.
Navarro JF, Milena FJ, Mora C, et al. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol 2006;26:562–570.
Acknowledgements
We thank Mar Gonzalez Garcia-Parreño for help with confocal microscopy and Susana Carrasco with histological studies. This work was supported by the grants FIS 06/0046, FIS PS09/00447, PI081564, EUproject DIALOK: LSHB-CT-2007-036644, ISCIII-RETIC REDinREN/RD06/0003, REDinREN/RD06/0004, Fondecyt (Chile) 1080083. Comunidad de Madrid/FRACM/S-BIO0283/2006, SAF 2007/63648 y CAM S-GEN-0247/2006. The salary support was provided by the FIS to MDSN, ABS, Programa Intensificación Actividad Investigadora (ISCIII/Agencia Lain-Entralgo/CM) to AO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Podocyte heat-shock protein 27 (HSPB1) is increased in diabetic nephropathy and focal segmental glomerulosclerosis. HSPB1 siRNA targeting increases apoptosis of podocytes sensitized to angiotensin II or in a high-glucose milieu. HSPB1 upregulation therefore allows podocytes to better withstand an adverse highglucose or angiotensin II-rich environment, such as found in diabetic nephropathy.
Supplementary information
Rights and permissions
About this article
Cite this article
Sanchez-Niño, M., Sanz, A., Sanchez-Lopez, E. et al. HSP27/HSPB1 as an adaptive podocyte antiapoptotic protein activated by high glucose and angiotensin II. Lab Invest 92, 32–45 (2012). https://doi.org/10.1038/labinvest.2011.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.138
Keywords
This article is cited by
-
Targeting the progression of chronic kidney disease
Nature Reviews Nephrology (2020)
-
Non-canonical NFκB activation promotes chemokine expression in podocytes
Scientific Reports (2016)
-
Albumin-induced apoptosis of tubular cells is modulated by BASP1
Cell Death & Disease (2015)
-
A combinatorial approach of Proteomics and Systems Biology in unravelling the mechanisms of acute kidney injury (AKI): involvement of NMDA receptor GRIN1 in murine AKI
BMC Systems Biology (2013)
-
3,4-DGE is cytotoxic and decreases HSP27/HSPB1 in podocytes
Archives of Toxicology (2013)
