
Abstract
The effect of diabetes mellitus vs the effect of the Ren2 gene on the glomerular pathology of (mREN-2)27 heterozygous male rats is controversial. As discrete diabetes-induced glomerular lesions may have been overlooked, we performed a detailed morphometric analysis of glomeruli in diabetic and non-diabetic heterozygous male (mREN-2)27 rats and their normotensive (non-diabetic and diabetic Sprague–Dawley) controls. Glomeruli were scored by light microscopy for nine discrete histological parameters, some of which were graded for extent and/or severity. Mesangiolysis, segmental hypocellularity, and severe tuft-to-capsule adhesions were specific to diabetes; severe mesangial matrix expansion, glomerulosclerosis, thickening of Bowman's capsule, and dilatation of the urinary space were specific to the Ren2 gene. Hyalinosis and hypercellularity were associated with both diabetes and the Ren2 gene: the effect was additive for hyalinosis and synergistic for hypercellularity. The histological parameters were then combined with two physiological indices (systolic blood pressure and proteinuria) and principle components analysis (PCA) was used to detect correlations between the variables. Four discrete patterns of pathology were identified; three were statistically associated with diabetes and/or the Ren2 gene. These findings suggest that both diabetes and the Ren2 gene make significant, albeit different, contributions to the glomerular pathology of diabetic heterozygous male (mREN-2)27 rats. Despite defining the contribution of diabetes, our work does not support the (mREN-2)27 rat as a model of diabetic nephropathy (DN). Rather, it suggests that these animals remain useful for investigating a particular and limited constellation of DN features.
Similar content being viewed by others


Main
There are numerous rodent models of human diabetic nephropathy (DN), attesting to the uncertainty associated with many of them.1, 2 Streptozotocin (STZ), which induces hyperglycemia by selectively damaging pancreatic β cells, has been administered to genetically modified rodents to create a variety of DN models.3, 4, 5, 6 The validity of one of these models, the diabetic (mREN-2)27 rat, has been questioned because of the apparent overwhelming impact of its genetically mediated hypertension on renal pathology.7 One group of investigators reported that STZ-induced diabetes in female heterozygous (mREN-2)27 rats recapitulates the proteinuria, nodular glomerulosclerosis, and progressive loss of renal function that are cardinal features of DN in human beings.6 However, other investigators suggest that the survival and kidney pathology of male heterozygotes is determined more by hypertension than by diabetes, as the presence of diabetes in these animals seemed to exert little phenotypic effect.7
A sexual dimorphism in hypertension is apparent from the lower blood pressure of female vs male (mREN-2)27 rats.8 This dimorphism has been proposed to underlie the apparent gender-dependent variation in experimental outcome because the effects of diabetes might only be observed in kidneys that are not dominated by hypertensive lesions.7 An equally plausible possibility, which is the focus of this study, is that the effect of diabetes in males has been overlooked because the types of renal damage typically assessed do not adequately capture the subtlety and extent of diabetic injury. As the (mREN-2)27 rat exhibits the elevated prorenin levels and high tissue renin–angiotensin system (RAS) activity that characterize DN in human beings,9, 10, 11, 12, 13, 14, 15, 16, 17 investigating this possibility is important lest a potentially valuable model be prematurely dismissed.
Light microscopy was used to examine the kidneys of diabetic male heterozygotes and their normotensive and non-diabetic controls for a wide variety of glomerular lesions not earlier assessed in the (mREN-2)27 rat. We also used principle components analysis (PCA) to identify discrete patterns of pathomorphology. We focused on the glomerulus alone as the pathological changes in many diseases progressing to chronic renal failure, including human and experimental DN, are initially apparent at the level of the glomerulus.18
MATERIALS AND METHODS
Animals
All animal experiments were carried out in accordance with guidelines set out by the Animal Ethical Committee Protocol C10/04, University of Otago, New Zealand. Breeding colonies of homozygous (mREN-2)27 and Sprague–Dawley Mol (SD; the wild-type strain from which the original transgenic founder was derived) rats were established using five breeding pairs of each strain (Taconic, Europe). (mREN-2)27 homozygous breeders were treated with 20 mg enalapril (Multichem, Auckland, New Zealand) per 700 ml drinking water. (mREN-2)27 homozygous males were crossed with SD females to produce (mREN-2)27 heterozygotes for experiments. All rats were housed in cages (3–4 animals per cage) in a room maintained at 22±2°C. They were exposed to a 12:12-h dark–light cycle and had unlimited access to tap water and standard rat chow (Weston Milling, Rangiora, New Zealand).
Induction and Monitoring of Diabetes
(mREN-2)27 heterozygotes and age-matched SD control rats were inducted into the study after having reached an average body weight of 200 g, which was at 6–8 weeks of age. They received 55 mg/kg STZ (AppliChem GmbH, Germany) diluted in 0.1 M sodium citrate buffer (pH 4.5) by tail vein injection under isoflurane anesthesia after an overnight fast. A glucometer (Accu-check Advantage II Glucose Monitor, Roche Diagnostics) was subsequently used to monitor blood glucose levels each week. STZ-treated rats received 1–4 units of insulin (Protophane, Novo Nordisk Pharmaceuticals) three times per week to reduce mortality and promote nutrition while maintaining blood glucose above 27 mmol/l.
In Vivo Monitoring of Animals
Ten rats from each of the following four groups were followed for 4 months: non-diabetic SD, diabetic SD, non-diabetic (mREN-2)27, and diabetic (mREN-2)27. Each individual rat was assessed every 4 weeks to obtain measurements of systolic blood pressure (SBP) and both plasma creatinine and urinary protein concentration. Briefly, SBP was recorded in conscious rats by tail-cuff sphygmomanometry (PowerLab System, AD Instruments). Blood from the tail vein was collected in a heparinized tube and the plasma obtained after separation was stored at −20°C for subsequent creatinine analysis. Rats were housed in metabolic cages (Techniplast, New South Wales, Australia) for 24 h with food and water ad libitum to measure both food and fluid intake and urinary output.
Determination of Plasma Creatinine, Urinary Protein, and Urinary Albumin
Plasma creatinine was measured by a protocol adapted from an earlier described high performance liquid chromatography (HPLC) method.19 Briefly, plasma samples were deproteinated by precipitation and centrifugation and the resultant supernatant was diluted in a 20:80 methanol:acetonitrile solution. The diluted supernatant was injected into an alumina HPLC column that had been pre-equilibrated in an alkaline methanol:acetonitrile mobile phase. A spectrum was extracted at 236 nm for peak area analysis. Urinary protein was measured using a modified Bradford dye-binding method (Bio-Rad Protein Assay; Bio-Rad Laboratories, CA, USA). Urinary albumin was determined by a double-antibody radioimmunoassay, as described earlier.20
Examination of Renal Histopathology
Tissue preparation and microscopy
Rats were anesthetized (Pentobarb 300; 60 mg/kg body wt i.p.; National Veterinary Supplies, Auckland) and the left kidney was removed, sectioned both longitudinally and transversely and fixed overnight in 10% buffered formalin at 4°C. Kidney pieces were subsequently embedded in paraffin and sectioned at 2 μm using a Leica RM2125 RT microtome. Sections were stained with periodic acid-Schiff (PAS) and 100 glomeruli from each rat were examined by light microscopy (BX50; Olympus, Christchurch, New Zealand). Individual glomeruli were digitally photographed at a magnification of × 400 using a Leica DFC 420 CCD camera. Histopathological assessment was performed without the knowledge of the experimental group to which each kidney belonged.
The scoring system
Glomeruli from 10 randomly selected rat kidney sections were initially assessed to give a list of repeatedly occurring pathomorphological characteristics. This list was subsequently used to assess sets of 100 glomeruli from each of another 10 randomly selected rat kidney sections to determine whether it could provide an adequate representation of the observed glomerular pathology. The list was expanded when certain pathomorphological characteristics could not be described by the existing criteria and the new list was again tested using 100 glomeruli from another 10 randomly selected kidney sections. The list was refined in this manner until the pathomorphological characteristics of any given glomerulus could be reasonably described by the selected criteria.
Morphological characteristics assessed
The degree of mesangial matrix expansion (MME; fibrillar and heterogeneous PAS-positive material) was subjectively graded on a scale of 0–4: grade 0=no expansion; grade 1=MME up to 25% of glomerular tuft area; grade 2=MME 25–50% of glomerular tuft area; grade 3=MME 50 to 75% of glomerular tuft area; and grade 4=MME 75–100% of glomerular tuft area. Glomerulosclerosis (distinct from MME; solid areas of homogeneous, PAS-positive material accompanied by complete capillary occlusion) was scored as being either global (affecting 100% of the glomerular tuft area) or segmental (affecting only part of the glomerular tuft, typically occupying 10–60% of its total area) in its extent and nodular in character where appropriate. Tuft-to-capsule adhesion (adherence of the glomerular tuft to Bowman's capsule (BC)) was rated on a scale of 0–3 where grade 0=normal; grade 1=adherence of <10% of the tuft to BC at one or more places; grade 2=adherence of 10–50% of the tuft to BC; and grade 3=adherence of >50% of the tuft to BC. Mesangiolysis, an attenuation or dissolution of the mesangial matrix, was recognized as a widening and incomplete staining of mesangial areas and a degeneration of mesangial cells. It is sometimes accompanied by capillary dilatation and was recorded as being either absent, present, or present with capillary dilatation. Hypercellularity (>30 cells/5 cm2) and hypocellularity (<15 cells/5 cm2) were described as being either global or segmental in extent. Cell type was not specified because of the difficulties in discriminating between mesangial, epithelial, and endothelial cells on PAS-stained sections. Hyalinosis (homogeneous and eosinophilic PAS-positive acellular masses located within the glomerular tuft or BC), thickening of BC (a deposition of pale pink and acellular fibrotic tissue on the inside of the basement membrane of BC), and dilatation of the urinary space (a widening of Bowman's space such that it occupies ≥25% of the total glomerular area) were recorded as being either present or absent.
Statistical Analyses
The data on each morphological and physiological variable were initially subjected to analysis of variance (ANOVA), with two-way ANOVA used for data with strain (SD/(mREN-2)27) and diabetes (presence/absence) as fixed factors and two-way repeated measures ANOVA if there were multiple measures per animal. Any significant main effects or interactions so identified were further explored using Fisher's Protected Least Significant Difference tests. PCA with Varimax rotation and Kaiser normalization was used to identify factors (components) among the morphological and physiological variables. A variable was considered to be an important feature of a factor if its absolute factor loading score was ≥0.4. The extracted components were analyzed using ANOVA. Data were transformed where necessary using the natural logarithm of either x, x+1, or √x. A P-value of <0.05 was taken to indicate statistical significance. All analyses were carried out using SPSS software (SPSS, Chicago, IL, USA).
RESULTS
Physiological Parameters and Mortality
The administration of STZ resulted in hyperglycemia in both (mREN-2)27 and SD rats and blood glucose levels were maintained above 27 mmol/l for the subsequent 4-month duration of the study. Physiological parameters, where possible, were assessed in each of the 40 rats at 0, 2, and 4 months after STZ induction.
There was an effect of diabetes on food consumption, water consumption, and urinary flow between 1 and 4 months (P<0.001 in each case), with diabetic rats exhibiting higher values than non-diabetic rats for all three parameters (Figure 1a–c). There was also an effect of strain on water consumption (P=0.048) and an interaction between strain and time on urinary flow (P=0.007), with water intake and urinary output being greater from 1 to 4 months and 1 to 3 months, respectively, in (mREN-2)27 vs SD rats (Figure 1a and b). An interaction between time, strain, and diabetes on absolute body weight change (P=0.008) was identified: diabetic rats gained weight over time at a much slower rate than their non-diabetic counterparts and non-diabetic (mREN-2)27 rats gained weight over time at a faster rate than non-diabetic SD rats (Figure 1d). An effect of diabetes on kidney weight (P<0.001) was revealed, with diabetic rats having larger kidneys than non-diabetic rats at the 4-month time point (Figure 1e). There was an elevation in the kidney weight:body weight ratio at the 4-month time point in diabetic vs non-diabetic rats (P<0.001; Figure 1f), which presumably reflects the opposing actions of diabetes on kidney and body weight. Plasma creatinine concentration increased with time (P<0.001), but was lower in diabetic vs non-diabetic rats (P<0.001; Figure 1g). Creatinine clearance was significantly increased in diabetic (mREN-2)27 rats between 1 and 3 months relative to the other groups (P<0.05), and then declined to similar values at 4 months (data not shown; JV Hill, G Findon, RJ Appelhoff, and ZH Endre; unpubished data). The significant decline between 3 and 4 months suggests that renal function in the diabetic (mREN-2)27 rats mimics the early increase in GFR that is observed in clinical DN, which is followed by a decrease in GFR with advancing disease and proteinuria. This suggests that at 4 months, the (mREN-2)27 rats have entered the late stage of diabetes from a functional perspective.

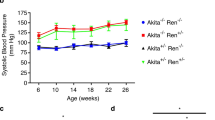
Physiological parameters in non-diabetic SD rats, diabetic SD rats, non-diabetic (mREN-2)27 rats, and diabetic (mREN-2)27 rats from 0 to 4 months. Data are presented as means±s.e.m. N=10 rats per group. P<0.05 for diabetic vs non-diabetic rats (*); P<0.05 for (mREN-2)27 vs SD rats (#); and P<0.05 for an interaction between strain and diabetes with post-hoc analysis indicating a difference between non-diabetic (mREN-2)27 rats and all other groups (R-). For (a–d) and (g–i): □=non-diabetic SD rats, ▪=diabetic SD rats, ○=non-diabetic (mREN-2)27 rats, and •=diabetic (mREN-2)27 rats. For (e) and (f): □=non-diabetic rats and ▪=diabetic rats. (a) Water consumption (ml/h); (b) urinary flow (ml/h); (c) food consumption (g/h); (d) absolute body weight change (g); (e) kidney weight (g); (f) kidney weight/body weight ratio; (g) plasma creatinine concentration (plasma [Cr]; μmol/l); (h) systolic blood pressure (SBP; mm Hg); and (i) urinary protein loss (mg protein/24 h).
There was an interaction between strain, diabetes, and time on SBP (P=0.002): SBP was significantly higher in (mREN-2)27 vs SD rats at all time points; diabetes increased SBP in SD rats at 2 and 4 months, but decreased it in (mREN-2)27 rats at 1, 3, and 4 months; and SBP in diabetic (mREN-2)27 rats increased between 0 and 2 and decreased between 3 and 4 months (Figure 1h).
An interaction between time and strain (P<0.001) and between time and diabetes (P=0.001) on urinary protein loss was identified: urinary protein loss increased in all rats between 0 and 1 month and remained constant thereafter, with this constant level of loss being greater for non-diabetic (mREN-2)27 rats than for all other groups (Figure 1i). Comparison of total urinary protein and urinary albumin in 16 4-month-old rats [4 SD, 8 (mREN-2)27, 4 diabetic (mREN-2)27] showed that total protein was largely representative of albuminuria in these groups: urinary protein was proportional to urinary albumin (r2=0.575; data not shown). However, proteinuria may underestimate albuminuria in some (mREN-2)27 rats as albumin made a greater contribution to total protein in the urine from three out of the eight (mREN-2)27 rats assayed. Exclusion of these rats from the linear regression analysis reduced the variability (r2=0.910; data not shown).
To avoid selection bias, mortality needs to be assessed in the context of the mortality within our larger rodent colony. A total of 156 male rats (30 non-diabetic SD; 39 diabetic SD; 45 non-diabetic (mREN-2)27; and 42 diabetic (mREN-2)27) have been monitored over a period of either 2 or 4 months. All rats were monitored after STZ induction, which took place at approximately 6–8 weeks of age. A total of six rats died unexpectedly during the designated monitoring period; five were non-diabetic (mREN-2)27 rats and one was a diabetic (mREN-2)27 rat. Death occurred between 2.1 and 5.5 months of age, with 6.7% of the non-diabetic (mREN-2)27 rats dying before the end of the 2-month monitoring period and a further 9.5% of the remaining non-diabetic (mREN-2)27 rats before the end of the 4-month monitoring period.
Renal Histopathology
The incidence of severe MME, urinary space dilatation, thickening of BC, glomerulosclerosis, mesangiolysis with and without capillary dilatation, tuft-to-capsule adhesions, hyalinosis, and both hypo- and hypercellularity differed between one or more of the individual groups of rats. These pathomorphological alterations are depicted in Figure 2.

Representations of the renal histopathological alterations whose incidence showed a statistically significant difference between non-diabetic SD rats, diabetic SD rats, non-diabetic (mREN-2)27 rats, and diabetic (mREN-2)27 rats. Original magnification: × 400. PAS-stained 2 μm paraffin sections. Abbreviations: TBC, thickening of Bowman's capsule; H, hyalinosis; SS, segmental sclerosis; M, mesangiolysis; CAP DIL, capillary dilatation. (a) Healthy glomerulus; (b) hypercellular glomerulus with severe MME; (c) glomerulus showing dilatation of the urinary space and thickening of Bowman's capsule; (d) globally sclerosed glomerulus with a thickened Bowman's capsule; (e) glomerulus showing segmental sclerosis, hyalinosis, and a moderate tuft-to-capsule adhesion; (f) glomerulus with segmental sclerosis that is nodular in character; and (g) mesangiolytic glomerulus displaying a severe tuft-to-capsule adhesion. Areas of matrix dissolution stain poorly with PAS and are characterized by a reduction in cell number that was scored as segmental hypocellularity. (h) Mesangiolytic glomerulus with several dilated capillary loops. (i) Mesangiolytic glomerulus with several dilated capillary loops, segmental sclerosis, an extensive area of hyalinosis, and a severe tuft-to-capsule adhesion.
Pathomorphology dependent on the Ren2 gene
Severe MME (MME score of 4) was significantly higher in (mREN-2)27 vs SD rats (P=0.042; Figure 3a). There was an interaction between strain and diabetes on the incidence of urinary space dilatation (P=0.002; Figure 3b), thickening of BC (P=0.007; Figure 3c), and glomerulosclerosis (P=0.004; Figure 3d), with non-diabetic (mREN-2)27 rats exhibiting higher incidences of these pathomorphological changes than all other groups. The incidence of segmental sclerosis as a variable per se (as opposed to glomerulosclerosis, which includes both global and segmental sclerosis) was too low to permit ANOVA. Nevertheless, sclerosis was nodular in character in 53.8% of the cases of segmental sclerosis, which itself occurred in 1–5% of the glomeruli in 12 (1 non-diabetic SD; 2 diabetic SD; 7 non-diabetic (mREN-2)27; and 2 diabetic (mREN-2)27) of the 40 rats. Overall, these data suggest that severe MME, urinary space dilatation, thickening of BC, and glomerulosclerosis are lesions that are primarily dependent on the presence of the Ren2 gene.

Incidence of severe MME, urinary space dilatation, thickening of Bowman's capsule, and glomerulosclerosis in non-diabetic SD rats, diabetic SD rats, non-diabetic (mREN-2)27 rats, and diabetic (mREN-2)27 rats at 4 months. Data are presented as means±s.e.m. N=10 rats per group. P<0.05 for (mREN-2)27 vs SD rats (#); and P<0.05 for an interaction between strain and diabetes with post hoc analysis indicating a difference between non-diabetic (mREN-2)27 rats and all other groups (R-). □=non-diabetic rats and ▪=diabetic rats. (a) Severe MME (square root transformation); (b) urinary space dilatation (square root transformation); (c) thickening of Bowman's capsule; and (d) glomerulosclerosis.
Pathomorphology dependent on diabetes
Mesangiolysis with capillary dilatation (Figure 4a), mesangiolysis without capillary dilatation (Figure 4b), segmental hypocellularity (Figure 4c), and severe tuft-to-capsule adhesions (those involving >50% of the glomerular tuft; Figure 4d) were significantly higher in diabetic vs non-diabetic rats (P=0.002, P=0.001, P=0.01, and P<0.001, respectively). An effect of strain, and not diabetes, on less severe tuft-to-capsule adhesions (those involving only 10–50% of the glomerular tuft) was identified: the Ren2 gene was associated with a significantly lower incidence (P=0.025) (Figure 4e). These data suggest that mesangiolysis, severe tuft-to-capsule adhesions, and segmental hypocellularity are primarily dependent on the presence of diabetes. They also suggest that the development of tuft-to-capsule adhesions was influenced by both diabetes and the Ren2 gene, with the former acting to promote, and the latter to antagonize, the generation of severe and moderate lesions, respectively.

Incidence of mesangiolysis (with and without capillary dilatation), segmental hypocellularity, and severe and moderate tuft-to-capsule adhesions in non-diabetic SD rats, diabetic SD rats, non-diabetic (mREN-2)27 rats, and diabetic (mREN-2)27 rats at 4 months. Data are presented as means±s.e.m. N=10 rats per group. P<0.05 for diabetic vs non-diabetic rats (*) and P<0.05 for (mREN-2)27 vs SD rats (#). □=non-diabetic rats and ▪=diabetic rats. (a) Mesangiolysis with capillary dilatation; (b) mesangiolysis without capillary dilatation; (c) segmental hypocellularity; (d) severe tuft-to-capsule adhesions; and (e) moderate tuft-to-capsule adhesions.
Pathomorphology dependent on both the Ren2 gene and diabetes
Hyalinosis was significantly higher in (mREN-2)27 than in SD rats (P<0.001) and in diabetic than in non-diabetic rats (P=0.005; Figure 5a). There was no interaction between strain and diabetes (P=0.174), which suggests that effects of diabetes and the Ren2 gene on hyalinosis were additive as opposed to synergistic.

Incidence of hyalinosis and hypercellularity in non-diabetic SD rats, diabetic SD rats, non-diabetic (mREN-2)27 rats, and diabetic (mREN-2)27 rats at 4 months. Data are presented as means±s.e.m. N=10 rats per group. P<0.05 for diabetic vs non-diabetic rats (*), P<0.05 for (mREN-2)27 vs SD rats (#), and P<0.05 for diabetic (mREN-2)27 vs diabetic SD rats (R+>SD+). □=non-diabetic rats and ▪=diabetic rats. (a) Hyalinosis and (b) hypercellularity.
Analysis of the combined data on global and segmental hypercellularity revealed an interaction between strain and diabetes (P=0.020): diabetic (mREN-2)27 rats had a greater number of glomeruli displaying global or segmental hypercellularity than diabetic SD rats, but this effect of strain was not apparent in the non-diabetic rats (Figure 5b). This synergism between strain and diabetes seemed to be due to diabetes increasing and decreasing cellularity in the presence and absence, respectively, of the Ren2 gene.
PCA analysis
The correlations between 13 of the morphological variables (MME, hypercellularity: global and segmental, hypocellularity: global and segmental, thickening of BC, urinary space dilatation, mesangiolysis with and without capillary dilatation, sclerosis: global and segmental, tuft-to-capsule adhesions involving >50% of the tuft, and hyalinosis) and 2 of the physiological variables (SBP and urinary protein loss) were explored using a PCA analysis to determine whether distinct patterns of histopathology could be identified. ANOVA was used to test the effects of strain and diabetes on the derived factors. A 4-factor solution to the PCA was selected, with the four factors cumulatively accounting for 72.7% of the total variance. The final communality estimates (ranging from 0.514 for tuft-to-capsule adhesions to 0.886 for segmental hypocellularity) indicated that much of the variance of the individual variables could be accounted for by these four factors (Table 1).
The first factor accounted for 21% of the total variance and was positively associated with high SBP, thickening of BC, urinary space dilatation, and both global and segmental glomerulosclerosis (Table 1). A two-way ANOVA of this factor revealed an interaction between strain and diabetes (P<0.001), with this pattern of pathology exhibiting a higher incidence in non-diabetic (mREN-2)27 rats than in all other groups (Figure 6a). The second factor accounted for 19.7% of the total variance and was positively associated with mesangiolysis (both with and without capillary dilatation), severe tuft-to-capsule adhesions, and segmental hypocellularity. It was also negatively associated with global hypercellularity (Table 1). A two-way ANOVA of this factor revealed a greater incidence in diabetic vs non-diabetic rat groups (P=0.002; Figure 6b). The third factor accounted for 18.1% of the total variance and was positively influenced by high MME scores and both global and segmental hypercellularity. It was also negatively associated with global hypocellularity (Table 1). A two-way ANOVA of this factor indicated strong trends toward an interaction between strain and diabetes (P=0.069), suggesting a higher level in diabetic (mREN-2)27 and a lower level in diabetic SD rat groups (Figure 6c). The fourth factor accounted for 13.8% of the total variance and was positively associated with hyalinosis, urinary protein loss, mesangiolysis with capillary dilatation, and segmental glomerulosclerosis (Table 1). A two-way ANOVA of this factor revealed a significantly higher incidence in (mREN-2)27 vs SD rat groups (P=0.047) and in diabetic vs non-diabetic rat groups (P=0.048; Figure 6d). Overall, these results suggest that four discrete patterns of pathology can be identified in our data set and that these patterns differentially associate with the Ren2 gene and diabetes.

Mean factor score for non-diabetic SD rats, diabetic SD rats, non-diabetic (mREN-2)27 rats, and diabetic (mREN-2)27 rats for PCA factors 1–4. Data are presented as means±s.e.m. N=10 rats per group. P<0.05 for an interaction between strain and diabetes with post hoc analysis indicating a difference between non-diabetic (mREN-2)27 rats and all other groups (R-), P<0.05 for diabetic vs non-diabetic rats (*), and P<0.05 for (mREN-2)27 vs SD rats (#). □=non-diabetic rats and ▪=diabetic rats. (a) Factor 1; positively influenced by high SBP, thickening of Bowman's capsule, urinary space dilatation, and both global and segmental glomerulosclerosis. (b) Factor 2; positively influenced by mesangiolysis without capillary dilatation, severe tuft-to-capsule adhesions, and segmental hypocellularity. (c) Factor 3; positively influenced by high MME scores and both global and segmental hypercellularity; negatively influenced by global hypocellularity. (d) Factor 4; positively influenced by hyalinosis, urinary protein loss, mesangiolysis with capillary dilatation, and segmental glomerulosclerosis.
DISCUSSION
This study has identified distinct as well as both additive and synergistic effects of the Ren2 gene and diabetes in male heterozygous (mREN-2)27 transgenic rats. Diabetes was associated with mesangiolysis, severe tuft-to-capsule adhesions, and segmental hypocellularity. In contrast, the Ren2 gene was associated with hypertension, thickening of BC, urinary space dilatation, and both global and segmental glomerulosclerosis. In addition to these discrete effects, our results suggest that diabetes and the Ren2 gene act additively to promote the development of a pattern of pathology characterized by hyalinosis, urinary protein loss, mesangiolysis with capillary dilatation, and segmental glomerulosclerosis. There was also a tendency for diabetes and the Ren2 gene to act synergistically to promote the development of a pattern characterized by high MME scores, hypercellularity (both global and segmental), and an absence of global hypocellularity, but the interaction between strain and diabetes did not quite reach statistical significance. Nevertheless, effects and interactions revealed by individual ANOVA of severe MME and hypercellularity supports the interaction suggested by PCA analysis. In this regard, although we acknowledge that the PCA needs to be interpreted with caution because of the number of cases available being limited for the inclusion of 15 variables, we note that there was generally a good correspondence between the ANOVA results of the PCA and those of the individual variables. Thus, when individually subjected to ANOVA, the variables that are correlated in the four PCA factors exhibited patterns of association with the Ren2 gene and diabetes that mimic those of the factors themselves.
Overall, these findings suggest that both diabetes and the Ren2 gene contribute to the glomerular pathology of diabetic male heterozygous (mREN-2)27 rats. The pathomorphological alterations that contribute to the pattern associated with the Ren2 gene have been documented in earlier morphometric studies of (mREN-2)27 rats.6, 10, 21 However, those that contribute to the pattern associated with diabetes have not been reported in similar studies of the diabetic (mREN-2)27 rat. In these animals, renal pathology has been reported to be far more dependent on hypertension than on diabetes, with the contribution of diabetes to glomerular pathology being limited to additive effects on MME.7 It is possible that the diabetic effects we observed have been hitherto overlooked because of the almost exclusive focus of earlier glomerular assessments of diabetic (mREN-2)27 rats on the incidence of glomerulosclerosis and degree of MME.6, 7 It is also possible that hypertensive lesions were much more prominent in one of the earlier renal histopathological assessments of diabetic (mREN-2)27 rats because of diabetes having been induced in rats that were likely to have been hypertensive for several weeks longer than those used in our study.7
Mesangiolysis was the most prominent diabetes-specific phenomenon identified in our study. At least three types of mesangiolysis have been identified to date. They differ with respect to their underlying cause and coexistent morphological features.22, 23 Mesangiolysis in this study was not accompanied by the gross capillary microaneurysms that characterize mesangiolysis resulting from a primary injury to either the mesangial matrix or mesangial cells. It may, therefore, more closely resemble either that occurring secondary to endothelial cell injury or that resulting from relatively mild but persistent damage to both endothelial and mesangial cells. Although further studies involving electron microscopy would be required to distinguish between these two possibilities, there is already a large body of research documenting hyperglycemia-induced perturbations in the biochemical processes carried out by both endothelial and mesangial cells.24, 25, 26, 27, 28 Some of these perturbations have been associated with mesangiolysis,4, 5, 23, 29, 30 which suggests that injury to both cell types has a pathogenetic function in the diabetes-associated mesangiolysis observed here. In addition, as matrix dissolution, which may result in loss of capillary wall anchor points in the mesangium, is thought to lead to capillary dilatation only if there is a concomitant loss of podocyte integrity,23, 31 it is possible that mesangiolysis with capillary dilatation also reflects diabetes-induced podocyte damage.
The association of the Ren2 gene with a pattern of pathology that correlates high SBP with thickening of BC, dilatation of the urinary space, and both global and segmental glomerulosclerosis suggests that these forms of Ren2 gene-dependent pathomorphology are mediated by hypertension. Ang II has a major function in the pathogenesis of hypertension in the (mREN-2)27 rat.9, 32, 33, 34 The observation that Ang II-induced glomerular injury can be prevented by protecting Ang II-infused kidneys from the increase in renal perfusion pressure35 is consistent with our suggestion that some of the pathological changes associated with the Ren2 gene may be mediated by hypertension. In addition, our study also suggests that some of the damage associated with the Ren2 gene may be hypertension independent. Urinary protein loss, hyalinosis, and some of the variance in segmental glomerulosclerosis were not correlated with hypertension in the PCA, despite ANOVA of these variables revealing clear effects of the Ren2 gene. This may be indicative of the non-hemodynamic actions of Ang II, activation of the Ang II-independent pathway of the receptor-associated prorenin system, or both, having a function in the development of these pathological alterations. However, as our experimental design did not attempt to separate hypertension-mediated Ren2 damage from hypertension-independent Ren2 damage, further experiments using RAS-independent blood pressure lowering agents would need to be performed to examine the precise function of hypertension in the pathomorphology of the (mREN-2)27 rat.
A caveat to our findings concerning the effects of the Ren2 gene is that genetic background differences have been found to affect the penetrance of the malignant hypertensive phenotype in (mREN-2)27 rats.36, 37, 38 As the original (mREN-2)27 transgenic founder was not generated on an inbred background, genetic background differences between the (mREN-2)27 heterozygotes and the parental SD controls could have contributed to the strain differences observed in this study.
Overall, our study suggests that both diabetes and the Ren2 gene contribute to the development of glomerular pathology in male diabetic (mREN-2)27 rats. It also suggests that two of the arguments put forward to discredit the diabetic (mREN-2)27 rat as a valid model of human DN are unfounded. The first of these is that the diabetic (mREN-2)27 rat does not recapitulate pathological changes, such as mesangiolysis, that are common in the later stages of DN.4 The second is that the presence of diabetes has little additional effect on the renal pathology of male (mREN-2)27 rats.7
Despite the rejection of such arguments, our work does not support the use of the male diabetic (mREN-2)27 rat as an exact model of human DN. Our histopathological assessment of the glomeruli from these animals rather suggests that they do not recapitulate all clinical manifestations of human DN. In particular, there was no worsening of mesangiolysis in diabetic (mREN-2)27 vs diabetic SD rats, or of glomerulosclerosis, thickening of BC, and urinary protein loss in diabetic (mREN-2)27 vs non-diabetic (mREN-2)27 rats. Furthermore, the non-diabetic (mREN-2)27 rats had a higher mortality than their diabetic counterparts, and the nodular sclerosis we observed was not reminiscent of the classical Kimmelstiel–Wilson pathology seen clinically. Nodules in human DN are often surrounded by a small rim of intact capillaries and frequently show a lamellated appearance, thought to result from repeated rounds of mesangiolysis followed by augmented extracellular matrix synthesis.39, 40, 41 Although capillary dilatation was associated with some nodular sclerosis in our study, we did not observe lamellation.
Although the male diabetic (mREN-2)27 rat does not fully recapitulate human DN, it may retain some usage in DN investigation because of its manifestation of a number of histopathological features (severe MME, mesangiolysis with and without capillary dilatation, and hyalinosis) that co-occur in the clinical condition. We draw attention to the increased prevalence of hyalinosis in diabetic (mREN-2)27 vs diabetic SD and non-diabetic (mREN-2)27 rats. Although not pathognomonic, this lesion is common in cases of human DN.41, 42 Investigations using the diabetic (mREN-2)27 rat may, therefore, be able to provide some insight into the molecular mechanisms and physiological perturbations underlying certain features of DN. This is important given that none of the currently available DN models perfectly reproduce the clinical condition.1, 2, 3, 4, 5, 6 Thus, rather than supporting the male diabetic (mREN-2)27 rat as a model of human DN, our work suggests that these animals might be useful for investigators studying a particular constellation of DN features.
References
Breyer MD, Bottinger E, Brosius III FC, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol 2005;16:27–45.
Tesch GH, Allen TJ . Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology (Carlton) 2007;12:261–266.
Lassila M, Seah KK, Allen TJ, et al. Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: role of advanced glycation end products. J Am Soc Nephrol 2004;15:2125–2138.
Nakagawa T, Sato W, Glushakova O, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 2007;18:539–550.
Kanetsuna Y, Takahashi K, Nagata M, et al. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol 2007;170:1473–1484.
Kelly DJ, Wilkinson-Berka JL, Allen TJ, et al. A new model of diabetic nephropathy with progressive renal impairment in the transgenic (mRen-2)27 rat (TGR). Kidney Int 1998;54:343–352.
Hartner A, Cordasic N, Klanke B, et al. Renal injury in streptozotocin-diabetic Ren2-transgenic rats is mainly dependent on hypertension, not on diabetes. Am J Physiol Renal Physiol 2007;292:F820–F827.
Langheinrich M, Lee MA, Bohm M, et al. The hypertensive Ren-2 transgenic rat TGR (mREN2)27 in hypertension research. Characteristics and functional aspects. Am J Hypertens 1996;9:506–512.
Mullins JJ, Peters J, Ganten D . Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature 1990;344:541–544.
Bachmann S, Peters J, Engler E, et al. Transgenic rats carrying the mouse renin gene—morphological characterization of a low-renin hypertension model. Kidney Int 1992;41:24–36.
Hilgers KF, Hartner A, Porst M, et al. Angiotensin II type 1 receptor blockade prevents lethal malignant hypertension: relation to kidney inflammation. Circulation 2001;104:1436–1440.
Hartner A, Porst M, Klanke B, et al. Angiotensin II formation in the kidney and nephrosclerosis in Ren-2 hypertensive rats. Nephrol Dial Transplant 2006;21:1778–1785.
Bohm M, Lee M, Kreutz R, et al. Angiotensin II receptor blockade in TGR(mREN2)27: effects of renin-angiotensin-system gene expression and cardiovascular functions. J Hypertens 1995;13:891–899.
Gurley SB, Coffman TM . The renin-angiotensin system and diabetic nephropathy. Semin Nephrol 2007;27:144–152.
Deinum J, Ronn B, Mathiesen E, et al. Increase in serum prorenin precedes onset of microalbuminuria in patients with insulin-dependent diabetes mellitus. Diabetologia 1999;42:1006–1010.
Hsueh WA, Anderson PW . Prorenin and the renin-angiotensin system in diabetic nephropathy. Semin Nephrol 1993;13:573–580.
Luetscher JA, Kraemer FB, Wilson DM, et al. Increased plasma inactive renin in diabetes mellitus. A marker of microvascular complications. N Engl J Med 1985;312:1412–1417.
Kriz W, LeHir M . Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int 2005;67:404–419.
Van Landuyt KG, Thienpont LM, De Leenheer AP, et al. Determination of creatinine in human serum using isocratic HPLC with an aluminum oxide stationary phase. J Chromatogr Sci 1994;32:294–297.
Jerums G, Allen TJ, Cooper ME . Triphasic changes in selectivity with increasing proteinuria in type 1 and type 2 diabetes. Diabet Med 1989;6:772–779.
Vernerova Z . [Morphological characteristic of renal injury in hypertensive rat strain transgenic for the mouse Ren-2 renin gene (TGR [mRen2] 27]. Cesk Fysiol 2007;56:79–84.
Morita T, Churg J . Mesangiolysis. Kidney Int 1983;24:1–9.
Morita T, Yamamoto T, Churg J . Mesangiolysis: an update. Am J Kidney Dis 1998;31:559–573.
Srinivasan S, Hatley ME, Bolick DT, et al. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 2004;47:1727–1734.
Xu B, Chibber R, Ruggiero D, et al. Impairment of vascular endothelial nitric oxide synthase activity by advanced glycation end products. FASEB J 2003;17:1289–1291.
Hink U, Li H, Mollnau H, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 2001;88:E14–E22.
Ha H, Lee HB . Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int Suppl 2000;77:S19–S25.
Ha H, Lee HB . Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology (Carlton) 2005;10:S7–S10.
Zhao HJ, Wang S, Cheng H, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 2006;17:2664–2669.
Stratta P, Canavese C, Mazzucco G, et al. Mesangiolysis and endothelial lesions due to peroxidative damage in rabbits. Nephron 1989;51: 250–256.
Kriz W, Gretz N, Lemley KV . Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998;54:687–697.
Bader M, Zhao Y, Sander M, et al. Role of tissue renin in the pathophysiology of hypertension in TGR(mREN2)27 rats. Hypertension 1992;19 (6 Part 2):681–686.
Moriguchi A, Brosnihan KB, Kumagai H, et al. Mechanisms of hypertension in transgenic rats expressing the mouse Ren-2 gene. Am J Physiol 1994;266 (4 Part 2):R1273–R1279.
Lo M, Medeiros IA, Mullins JJ, et al. High blood pressure maintenance in transgenic mRen-2 vs Lyon genetically hypertensive rats. Am J Physiol 1993;265 (1 Part 2):R180–R186.
Mori T, Cowley Jr AW . Role of pressure in angiotensin II-induced renal injury: chronic servo-control of renal perfusion pressure in rats. Hypertension 2004;43:752–759.
Whitworth CE, Fleming S, Cumming AD, et al. Spontaneous development of malignant phase hypertension in transgenic Ren-2 rats. Kidney Int 1994;46:1528–1532.
Whitworth CE, Fleming S, Kotelevtsev Y, et al. A genetic model of malignant phase hypertension in rats. Kidney Int 1995;47:529–535.
Kantachuvesiri S, Haley CS, Fleming S, et al. Genetic mapping of modifier loci affecting malignant hypertension in TGRmRen2 rats. Kidney Int 1999;56:414–420.
Saito Y, Kida H, Takeda S, et al. Mesangiolysis in diabetic glomeruli: its role in the formation of nodular lesions. Kidney Int 1988;34:389–396.
Stout LC, Kumar S, Whorton EB . Focal mesangiolysis and the pathogenesis of the Kimmelstiel-Wilson nodule. Hum Pathol 1993;24:77–89.
Fogo AB, Kashgarian M, Brenner BMK . Diagnostic Atlas of Renal Pathology: A Companion to Brenner & Rector's the Kidney, 7th edn. Elsevier Saunders: Philadelphia, PA [Edinburgh], 2005, viii, 480 pp.
Alsaad KO, Herzenberg AM . Distinguishing diabetic nephropathy from other causes of glomerulosclerosis: an update. J Clin Pathol 2007;60:18–26.
Acknowledgements
We thank Estelle Fraser and Amanda Fahy for technical assistance with embedding and sectioning, Associate Professor John Leader, Dr Jennifer Bedford, and Dr Alastair Murray for advice on histopathological examination and scoring, Professor Glenda Gobe and Professor Agnes Fogo for critical reading of the manuscript, and the late Dr Helen Milne for helpful discussions. This study was supported by grants from New Zealand National Heart Foundation (#1201), New Zealand Kidney Foundation (#11), Lottery Health Research New Zealand (#207532), University of Otago, Diabetes Christchurch Incorporated, Diabetes Training and Research Trust, and Canterbury Medical Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Appelhoff, R., Hill, J., Findon, G. et al. Differential contribution of diabetes and the Ren2 gene to glomerular pathology in diabetic (mREN-2)27 rats. Lab Invest 90, 1225–1235 (2010). https://doi.org/10.1038/labinvest.2010.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.96
