
Abstract
Human sarcomas are a heterogeneous group of over 50 different malignant tumors for which very few diagnostic markers currently exist. MicroRNA (miRNA) transcript levels have been proposed for use in the diagnosis, classification and prognosis of tumors. Over 700 miRNAs are identified in humans and miRNA are considered attractive candidates for developing novel biomarkers in sarcomas. However, miRNA expression patterns found in sarcomas are poorly understood and no central resource exists to contain this information. To systematically address the gap in both biological knowledge and bioinformatics infrastructure, we generated miRNA expression profiles for over 300 tumor tissue samples representing 22 different sarcoma types and developed a web-accessible database to enable facile access to the data. Sarcoma microRNA Expression Database (S-MED) is a repository that describes the patterns of miRNA expression in various human sarcoma types. S-MED provides both basic and advanced data search options for exploration of the data in heat map and text/numerical formats. The database also provides statistical details such as fold changes and P-values for differentially expressed miRNAs in each sarcoma type and corresponding normal tissue. Further, we have experimentally validated differentially expressed miRNAs in angiosarcoma and other sarcoma types. This comprehensive database is the first of its kind specifically devoted to miRNA expression patterns in sarcomas is available through the URL link http://www.oncomir.umn.edu/.
Similar content being viewed by others


Main
Sarcomas are a highly heterogeneous group of aggressive and invasive mesenchymal tumors. Over 50 histological subtypes exist and their diagnostic classification is a major challenge.1 Sarcomas can be found throughout the body but the majority of them occur in the extremities. Many types are named after the type of cell, or tissue involved, as in angiosarcoma, chondrosarcoma, fibrosarcoma, liposarcoma and osteosarcoma. Sarcoma tumors can also be of pleomorphic subtypes of cryptic origins, such as malignant fibrous histiocytoma (MFH). Sarcomas are associated with a significant risk of developing distant metastasis depending on the tumor type and grade.
MicroRNAs (miRNAs) are a recently identified class of small noncoding RNAs that have been proposed for use in the diagnosis, classification and prognosis of tumors.2 More than 700 miRNAs are identified in humans,3 and they are attractive candidates for developing novel diagnostic and prognostic markers for a wide variety of sarcomas. The miRNAs negatively regulate gene expression by binding to the 3′ UTRs of messenger RNA.4 Extensive study of miRNA biogenesis and computational analysis of miRNA target genes has resulted in the development of miRNA-related target prediction software and predicted target database.5, 6, 7 Furthermore, miRNAs deregulated in various human diseases have been cataloged.8 Analysis of miRNA expression has been extensively carried out in carcinomas and other hematological tumors.9, 10, 11, 12, 13 However, there is no comprehensive expression analysis that catalogs miRNA expression patterns in the wide variety of existing sarcoma types with sufficient sample size to carry statistical power. In our earlier studies, we have reported preliminary analysis of miRNA expression levels in a small series of sarcomas14 as well as in colon cancer.15
Exhaustive analysis of miRNA expression patterns in sarcomas will accelerate the identification and development of novel miRNA-based biomarkers for these tumors. To achieve this goal, we have created a comprehensive database containing miRNA expression patterns for 22 different types of human sarcomas and corresponding normal tissue types.
MATERIALS AND METHODS
Tumor Samples
Snap-frozen patient tumor tissue samples were collected through the tissue procurement facility and from our collaborators at the Masonic Cancer Center, University of Minnesota, and stored at −80°C until used. Institutional review board approved this study.
miRNA Expression Profiling
miRNA was isolated using the miRvana RNA isolation kit (Ambion, Austin, TX, USA) following the manufacturer's protocol. RNA was quantified using a Nanodrop spectrophotometer (Theromo Fisher, Wilmington, DE, USA) and quality was determined by denaturing agarose gel electrophoresis as well as on Agilent 6000 nanochip (Agilent Technologies, Palo Alto, CA, USA). Only samples with RNA index number (RIN) of ⩾5 were subjected to miRNA analysis using miRNA BeadArrays (Illumina, San Diego, CA, USA). The miRNA BeadArray procedure is similar to the cDNA-mediated annealing, selection, extension and ligation (DASL) method reported earlier.16 After hybridization, the arrays were imaged using an Illumina BeadArray Reader and the fluorescent intensity of miRNA probes were analyzed using BeadStudio version 3.1.1(Illumina).
Data Analyses
Raw miRNA expression data were quantile normalized17 on a phase-by-phase basis. Differential expression was analyzed using a two-group t-test to determine miRNAs that were differentially expressed between a specific sarcoma and the other sarcomas included in a particular phase of analysis. When analyzing tumors types from different phases, for use in the advanced search, expression data from all phases were once again quantile normalized together. Quantile normalization, unsupervised hierarchical clustering and statistical analyses were carried out using GeneData Expressionist Software (Genedata, San Francisco, CA, USA).
Web Application
The application was created using the LAMP software bundle (Linux, Apache2, MySQL 5.0 and PHP) and HTML. The entire application is hosted on the web for researchers across the globe to access and review the information stored in the data set. An Apache web server is used to host the web application. PHP is used to generate the user interface and used to communicate with the MySQL database at the backend. Normalized expression data, statistical results and annotation data are stored within the database. A user-friendly graphical interface is provided to assist the users for ease of data retrieval and for selection of different criteria for data analysis. PHP is used to generate the HTML content through a database-driven architecture that was designed for incorporation of additional information.
Quantitative Real-Time PCR Analysis
RNAs isolated from patient tissues were reverse transcribed and PCR amplified with miScript reverse transcription kit and miScript SYBR PCR kit (Qiagen, Valencia, CA, USA) using ABI 7500 real-time PCR system following the manufacturer's protocols. Values from duplicate reactions were averaged and normalized to the level of U6 SnoRNA. Relative expression levels were calculated following comparative Ct method.18
RESULTS
Data Content
Sarcoma microRNA Expression Database (S-MED) is a comprehensive repository that contains the patterns of miRNA expression found in over 300 sarcoma samples representing 22 tumor types and select normal tissue samples (Table 1). The web-accessible interface (http://www.oncomir.umn.edu) allows users to determine the transcript level of specific miRNAs of interest contained within the database. S-MED provides both ‘Basic search’ and ‘Advanced search’ options for exploration and visualization of the data in heat map and numerical formats. The database also provides statistical details such as fold changes and P-values for differentially expressed miRNAs in each sarcoma and normal tissue type.
Accessing the Database
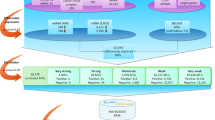
‘Basic search’ allows the retrieval of the expression level for a specific miRNA of interest (Figure 1a). Selecting one or multiple miRNA from the list of all miRNA available in the database returns a heat map sorted by sarcoma type and experimental phase (Figure 1b). The initial result returned to the user provides the data in heat map form based on the overall intensity of the signal in which the value returned is represented by a color ranging from black to blue to yellow with a range of values from <250 to >32000. A key is provided in the upper right hand corner of the data view. The user is then given the additional options of seeing the data in numerical format or as a relative intensity heat map (Figures 1c and d). The relative intensity heat map shows the relative expression level in fold change relative to the average across the phase range of normalized values from 0.125- to 8-fold. miRNA, which are decreased relative to the average, are shown in progressive more intense green and miRNA, which show increased levels relative to the average, are shown in progressively more intense red. Each of the phases (sets of experiments carried out simultaneously) are both normalized and averaged separately in basic search to allow for future integration of unrelated data sets and additional platform types such as direct sequencing. In all, 735 miRNAs were analyzed in the first phase, whereas 1146 miRNAs were analyzed in the second, third and fourth phases. Should a miRNA not be present in a particular phase or group of the analysis, no data will be shown for the group or phase.

Screen shots of information flow in S-MED. Flowchart shows an example of information retrieval from S-MED. Arrows indicate where a click will lead within the web application. (a) Initial basic search selection option. Selection of one or more miRNA and clicking on the submit button will transfer the user to the screen shown in panel b. (b) Absolute intensity data view. From here the user can navigate to view the numeric data used to generate the heat map by clicking ‘Show numeric data’ link, the relative intensity view by clicking ‘Show relative data view’, or the database and annotation page by clicking on the name of the miRNA as shown by the arrows. (c) Absolute data view in text format. (d) Relative intensity data view. (e) Database annotation and external links.
Clicking on the miRNA name in any of the expression tables will take the user to a page that describes the associated annotation such as miRNA name, Illumina probe ID, chromosomal location, coordinates (if a single genomic location is available) and sequences of the probe (Figure 1e).
In addition, the user can return to the ‘expression data’ views for the probe, statistical data as discussed below, predicted mRNA 3′ UTR targets available at miRDB,19 as well as a Google Scholar literature search keyed by the name of the miRNA. As an added feature, by clicking on ‘colocalized miRNA’, the user can visualize and analyze the expression patterns for miRNA found within common chromosomal regions.
‘Advanced search’ allows the user to directly define both the miRNA of interest and tissue types and examine direct comparisons of the data. Similar to the basic search, absolute, relative and numerical view options are also available for the selected data (Figure 2a). Uniqueness of advanced search is that, the data have been quantile normalized across all data sets, and the selected data are available on the same x axis. Importantly, only the data selected are used to generate the relative view allowing for direct visualization of differential expression between selected tissue types. In both basic and advanced modes, relative values are not stored in the data repository, but are computed as and when the user desires to visualize certain cases against each other.

Screen shots of advanced search and statistical search. (a) Advanced search options allow the user to define miRNA and tissues of interest by visualizing on a single experimental axis. (b) Statistical result search starting from an miRNA of interest. This option allows the user to determine which tissue samples show changes in an miRNA of interest by considering the P-value and or the fold change for a tissue relative to others in an experimental phase. (c) Statistical result search starting with a specific tissue of interest. This access option allows the user to define a statistical threshold, a tissue type of interest and a fold change to identify miRNA that are of potential interest.
Statistical results are also included in S-MED. The user can select a miRNA and obtain all the statistical results for that miRNA that are contained within the statistical results table of the database. Following selection, an html table is generated, which provides the tissue type, phase, P-value for a two group t-test comparing all tissue types available in a particular experimental phase and the average fold change for the relevant miRNA (Figure 2b).
For additional statistical function, the user can select a tissue type, a statistical threshold and fold change cutoff to return all miRNA that are differentially expressed in a specific tissue type with the cutoffs defined by the user. A table is then returned that provides tissue comparison, phase of the experiment, miRNA, P-value for a two group t-test between the selected tissue and all other tissues within the phase, and fold change between the average of the selected tissue and other experiments included in the phase, sorted by the magnitude of the fold change (Figure 2c). Heat maps for each of these miRNA individually can be examined by clicking on the name of the miRNA within the table or the entire set of miRNA by clicking on the ‘RNA expression’ link provided at the top of the page. Once the user has navigated to the expression view, all three methods of visualizing the data, relative, absolute or as text, can then be generated on demand. The data maintain the fold change sort from the statistical view within the heat maps. Except for MFH (high-grade undifferentiated pleomorphic sarcoma, not otherwise specified), each of the different sarcomas and normal tissue exhibit distinct and characteristic miRNA signatures that are significant beyond the Bonferroni correction.
Additional evidence attesting to the quality of the data can be obtained by examining unsupervised hierarchical clustering of the data. Average linkage correlation-based unsupervised clustering of all data, available for each phase mean centered by phases, reveals that the vast majority of the tissue profiles cluster based on the tumor type. (Figure 3) Several specific examples exist within the data, in which tumors of the same type clustered in separate groups. For example, in phase 1, the MFH are found in three distinct groups. In addition, examples exist in which single samples cluster within groups made predominantly of a different specific tumor type. For example, one of the osteosarcoma samples clustered within a group primarily derived from MFH tumors.

Unsupervised hierarchical clustering analysis of miRNA expression profiles in various sarcoma types. Average linkage correlation-based unsupervised hierarchical clustering was carried out on each phase using all expression data available for each phase. Branch points closer to the bottom of the tree correspond to higher correlations between profiles. (a) Phase 1. (b) Phase 2. (c) Phase 3. (d) Phase 4. It can be noted that all the miRNA expression data points are used in the generation of these dendrograms; a tighter clustering patterns may be obtained by further filtering the miRNAs.
To provide a useful case to demonstrate the utility and quality of both the data contained within this database and the database interface, we focused on examining and verifying the information contained within S-MED for angiosarcoma. Soft tissue angiosarcomas are malignancies showing endothelial differentiation and are highly aggressive. They vary in appearance and at extremes may resemble fibrosarcomas or undifferentiated carcinomas. To begin, we searched the S-MED statistical results by tissue group selecting only miRNA that met the Bonferroni correction to the multiple testing problem associated with microarray statistical analysis, tissue-type ‘angiosarcoma’ and miRNA that showed a >10-fold change. In total, 41 miRNA were identified and exhibited close chromosomal proximity in a cluster on chromosome 19. Examining the absolute values in heat map format revealed that, for the most part, these showed low expression levels across all the other sarcoma types and other tissues studied, with the exception of angiosarcoma. The ‘relative data’ view also showed this unique pattern of miRNA expression in angiosarcoma. It is interesting that one of the MFH tumors also showed very high levels of the chromosome 19 miRNA cluster.
In previous work, we have shown high correlations between miRNA levels measured by Illumina array profiling and quantitative real-time PCR (qRT-PCR).15 To further validate the unique expression pattern seen in angiosarcomas for chromosome 19 miRNA, we quantified the level of the differentially expressed miRNAs in selected tumor samples using qRT-PCR. qRT-PCR results confirmed the overexpression of miRNAs miR-515-3p, miR-515-5p, miR-517a, miR-517c, miR-518b, miR-519a and miR-522 in angiosarcoma relative to other sarcoma types (gastrointestinal stromal tumors, MFH and epithelioid sarcoma) included in the analysis (Figure 4). miR-515-3p and miR-517c are expressed over 12-fold in angiosarcoma relative to other sarcomas (Supplementary Figure 1). These findings suggested that miR-515-3p and miR-517c can be investigated as potential diagnostic markers for angiosarcoma.

(a) Heat map of absolute values for selected miRNA derived from angiosarcoma, GIST, MFH and epithelioid sarcoma. (b) Heat map of relative values contained in the database for selected miRNAs and tissues from a. (c) Quantitative PCR validation of expression of selected miRNA in GIST, MFH, angiosarcoma and epithelioid sarcoma. Ct values from two independent experiments per sample were normalized to the endogenous levels of U6. Two samples were tested for each sarcoma subtype and average values were shown for each sarcoma type. Error bars show the s.d. in two different samples per tested subtype.
DISCUSSION
Sarcomas are rare tumors and the availability of good quality, frozen tumor tissue samples is a major challenge in studying their gene/miRNA expression patterns. Availability of high quality, easily accessible miRNA expression data in the public domain from a significant number of sarcoma samples should accelerate the comprehension of data, which in turn should aid in better understanding of the biology, diagnosis and treatment of sarcomas. Recently, miRNA functional analyses of rhabdomysosarcoma (RMS) identified several miRNAs that are associated with RMS development and progression.20, 21 miRNAs such as miR-2920 and mir-20621 have significant role in muscle cell differentiation and deregulation of these miRs is implicated in RMS. Further, mutation and location-dependent miRNA expression patterns in gastrointestinal stromal tumors has been identified.22 However, a comprehensive miRNA expression database that will serve as a resource for comparing various sarcomas is still lacking. S-MED contains the expression patterns of miRNAs for 310 tissue samples, which represent 22 different sarcoma types. In addition, the database also contains miRNA expression data from normal tissues such as bone, smooth muscle and skeletal muscle allowing for comparison of sarcoma tissues with respective normal tissues. We have also included benign tumors such as neurofibroma and leiomyoma for comparative studies with their malignant counterparts. The presence of unique expression patterns among sarcoma types suggests that miRNAs may have a significant role in their onset and progression. Further, these miRNA signatures can be explored to identify diagnostic biomarkers and also to identify novel therapeutic targets. To this end, we have identified miRNAs, which are specifically overexpressed and downregulated in specific sarcoma types. The miRNA expression signatures from 22 different sarcoma types that are made available through S-MED will now serve as an important resource to (1) understand sarcoma biology, (2) develop novel miRNA-based biomarkers and (3) provide improved medical treatment of these rare and diagnostically challenging diseases.
Comprehending the numerical values in either absolute or normalized format is difficult. To aid interpretation and analysis, the option of visualizing the data as a heat map is provided. Different color codes are used to represent data in absolute and relative format. Further, user-defined data processing is possible by starting with the text values obtainable through the user interface. An advantage of this data repository is that instead of providing previously analyzed relative data to the end user, the database allows normalization of expression data for the selected sarcoma types. This allows the user to define the relationships they are interested, making data comprehension easier. An additional advantage is that, in our database, the statistical results are tightly integrated with the data that generated them.
In addition to generating the vast amount of expression data and making it available, we have also experimentally validated unique miRNA expression levels observed in angiosarcoma. Angiosarcoma showed unique miRNA expression patterns compared with other sarcoma types included in the analysis. We have validated this unique expression pattern observed in our miRNA microarray approach using qRT-PCR. Thus, we believe that the differential expression data has significant biological relevance.
Chromosome 19 miRNAs are one of the largest clusters ever reported and expressed at a high level in the placenta.23 It is not entirely surprising then to find them expressed at a high level in tumors associated with vessel formation. The rationale for why chromosome 19 miRNA are found at such a high level in angiosarcomas is beyond the scope of this paper. However, their expression in the placenta and in angiosarcoma is consistent with a potential role in regulating vessel growth, which may allow the sarcoma to grow unchecked.
One of the tumors labeled as MFH showed a high level of expression of miRNA specific to angiosarcoma. It is likely that this may represent an angiosarcoma mislabeled in the database. Owing to the complexity of sarcomas, molecular diagnostic separation through miRNA expression levels of tumors with similar histological indicators is an important next step in this study.
Both unsupervised hierarchical clustering and the statistical analyses show that unique patterns exist in the miRNA expression profiles obtained from various sarcoma types. The unsupervised hierarchical clustering patterns suggest (1) that additional molecular subtypes as defined by miRNA expression patterns may exist for several of the sarcoma types currently defined as single diagnostic entity based on histological features, and (2) that some of the sarcoma specimens may be misdiagnosed, which need further molecular diagnostic verifications. Further understanding of the relationship of these molecularly defined subtypes with patient treatment and outcomes is an important next step of this study.
We are currently working toward the understanding of the biological relevance of these expression patterns to sarcomagenesis. For instance, in osteosarcomas, we noticed significant downregulation of miRNAs, which were associated with chromosome 14 (Venugopal Thayanithy, unpublished observation), whereas in rhabdomyosarcomas, we observed differential expression of miRNA involved in muscle differentiation (Aaron L. Sarver, unpublished). In both these sarcomas, mRNA profiles obtained from identical tumors show tight correlations with the miRNA profiles.
In conclusion, we have compiled a comprehensive miRNA expression database for a variety of human sarcoma types. We believe this database will be a valuable resource for the academic community working on sarcoma and other tumor types, and should allow users to identify and develop novel biomarkers for various sarcoma types. In addition, our database should allow researchers in the solid tumor community (breast, colon and prostate) to determine the specificity of their potential miRNA biomarkers at early stages of research and better understand the common mechanisms of tumorigenesis in a wide variety of samples. Improved understanding of the molecular basis underlying sarcomagenesis will hasten the identification of novel therapeutic targets and decipher the signaling pathways that are affected in these sets of tumors. In the future, we will expand this resource to incorporate miRNA expression data from various sarcoma mouse models, which will promote comparative genomic studies by allowing the direct comparison of human tumor tissue data with mouse tumor tissue data to identify the most relevant mouse models for studying human sarcoma biology and treatment.
References
Weiss S, Goldblum J . Soft tissue tumors. Elsevier: Mosby, St Louis, 2008.
Calin GA, Croce CM . MicroRNA signatures in human cancers. Nat Rev Cancer 2006;6:857–866.
Griffiths-Jones S, Saini HK, van Dongen S, et al. miRBase: tools for microRNA genomics. Nucleic Acids Res 2008;36 (Database issue):D154–D158.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–233.
Xiao F, Zuo Z, Cai G, et al. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 2009;37 (Database issue):D105–D110.
Lewis BP, Shih IH, Jones-Rhoades MW, et al. Prediction of mammalian microRNA targets. Cell 2003;115:787–798.
Sethupathy P, Corda B, Hatzigeorgiou AG . TarBase: a comprehensive database of experimentally supported animal microRNA targets. RNA 2006;12:192–197.
Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 2009;37 (Database issue):D98–D104.
Calin GA, Ferracin M, Cimmino A, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005;353:1793–1801.
Calin GA, Liu CG, Ferracin M, et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 2007;12:215–229.
Ma L, Teruya-Feldstein J, Weinberg RA . Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007;449:682–688.
Takagi T, Iio A, Nakagawa Y, et al. Decreased expression of microRNA-143 and -145 in human gastric cancers. Oncology 2009;77:12–21.
Schetter AJ, Leung SY, Sohn JJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008;299:425–436.
Subramanian S, Lui WO, Lee CH, et al. MicroRNA expression signature of human sarcomas. Oncogene 2008;27:2015–2026.
Sarver AL, French A, Borralho PM, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer 2009;9:401.
Chen J, Lozach J, Garcia EW, et al. Highly sensitive and specific microRNA expression profiling using BeadArray technology. Nucleic Acids Res 2008;36:e87.
Bolstad BM, Irizarry RA, Astrand M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003;19:185–193.
Schmittgen TD, Livak KJ . Analyzing real-time PCR data by the comparative Ct method. Nat Protoc 2008;3:1101–1108.
Wang X . miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA 2008;14:1012–1017.
Wang H, Garzon R, Sun H, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008;14:369–381.
Taulli R, Bersani F, Foglizzo V, et al. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J Clin Invest 2009;119:2366–2378.
Haller F, von Heydebreck A, Zhang JD, et al. Localization- and mutation-dependent microRNA (miRNA) expression signatures in gastrointestinal stromal tumours (GISTs), with a cluster of co-expressed miRNAs located at 14q32.31. J Pathol 2009;220:71–86.
Bentwich I, Avniel A, Karov Y, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 2005;37:766–770.
Acknowledgements
We thank Drs Andrew Fire, Clifford Steer, Jaime Modiano, Lei Li, Matt van de Rijn and Reena Kartha for their comments and suggestions. We also thank Dr Amy Skubitz and Sarah Bowell of the tissue procurement facility for their help. The S-MED was developed with funding from Karen Wyckoff Sarcoma Foundation, Department of Laboratory Medicine and Pathology, and Academic Health Center, University of Minnesota, Minneapolis. We also acknowledge the Minnesota Supercomputing Institute for providing extensive computational resources (hardware and systems administration support). The S-MED database is available through the URL link http://www.oncomir.umn.edu/
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Supplementary information
Rights and permissions
About this article
Cite this article
Sarver, A., Phalak, R., Thayanithy, V. et al. S-MED: Sarcoma microRNA Expression Database. Lab Invest 90, 753–761 (2010). https://doi.org/10.1038/labinvest.2010.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.53
Keywords
This article is cited by
-
miRNA deregulation targets specific pathways in leiomyosarcoma development: an in silico analysis
Journal of Translational Medicine (2019)
-
Potent and PPARα-independent anti-proliferative action of the hypolipidemic drug fenofibrate in VEGF-dependent angiosarcomas in vitro
Scientific Reports (2019)
-
OMCD: OncomiR Cancer Database
BMC Cancer (2018)
-
Biological analysis of cancer specific microRNAs on function modeling in osteosarcoma
Scientific Reports (2017)
-
microRNAs Databases: Developmental Methodologies, Structural and Functional Annotations
Interdisciplinary Sciences: Computational Life Sciences (2017)

{kind=link}