
Abstract
Hepatic sinusoidal endothelial cells (HSECs) are a unique subpopulation of fenestrated endothelial cells lining the hepatic sinusoids and comprising the majority of endothelial cells within the liver. HSECs not only have important roles in blood clearance, vascular tone, and immunity, but also undergo pathological changes, contributing to fibrosis, angiogenesis, and portal hypertension. There are few cell culture models for in vitro studies of motility and angiogenesis as primary cells are time-consuming to isolate, are limited in number, and often lack features of pathological vasculature. The aim of this study was to generate an immortalized cell line derived from HSECs that mimic pathological vasculature and allows detailed molecular interventions to be pursued. HSECs were isolated from mouse liver using CD31-based immunomagnetic separation, immortalized with SV40 large T-antigen, and subcloned on the basis of their ability to endocytose the acetylated low-density lipoprotein (AcLDL). The resulting cell line, transformed sinusoidal endothelial cells (TSECs), maintains an endothelial phenotype as well as some HSEC-specific features. This is evidenced by typical microscopic features of endothelia, including formation of lamellipodia and filopodia, and a cobblestone morphology of cell monolayers. Electron microscopy showed maintenance of a limited number of fenestrae organized in sieve plates. TSECs express numerous endothelia-specific markers, including CD31 and von Willebrand's factor (vWF), as detected by PCR array, immunoblotting, and immunofluorescence (IF). Functionally, TSECs maintain a number of key endothelial features, including migration in response to angiogenic factors, formation of vascular tubes, endocytosis of AcLDL, and remodeling of extracellular matrix. Their phenotype most closely resembles the pathological neovasculature associated with chronic liver disease, in which cells become proliferative, defenestrated, and angiogenic. Importantly, the cells can be transduced efficiently with viral vectors. TSECs should provide a reproducible cell culture model for high-throughput in vitro studies pertaining to a broad range of liver endothelial cell functions, but likely broader endothelial cell biology as well.
Similar content being viewed by others


Main
Hepatic sinusoidal endothelial cells (HSECs) are a morphologically and functionally unique sub-population of liver endothelial cells that form the lining of the hepatic sinusoids. These cells comprise the vast majority of endothelial cells within the liver, but differ dramatically from endothelia of other organs in that they contain numerous fenestrations and lack a basement membrane.1 Recent intensive study of HSECs continues to expand our understanding of these cells and reveals their role in a diverse array of homeostatic functions in the liver, including clearance of waste products from the blood, regulation of pericyte contractility, and innate immune function.2
Not only are HSECs unique in their normal structure and function, but also in their contribution and response to liver pathology. Chronic liver injury and cirrhosis are associated with a robust angiogenic response, with the formation of a dense neovasculature in the fibrotic septa surrounding regenerative nodules.3, 4 These pathological vessels become capillarized and defenestrated, and form a more classic vascular basement membrane.5 Liver endothelial cells in these circumstances take on an ‘activated’ angiogenic phenotype, which includes altered surface markers6 and changes in both morphology and behavior,7 allowing increased proliferation and angiogenic invasion. Similar changes can be seen in the settings of portal hypertension,8 hepatocellular carcinoma9 and during the aging process.10
Although the above concepts represent significant advances in our understanding of the physiology and pathophysiology of the unique endothelium within the liver, many aspects of the biology of these cells remain poorly understood, owing, in part, to the relative paucity of appropriate in vitro models. The development of several methods to isolate liver endothelial cells from experimental animals,11, 12, 13 although a significant and critically important advancement, still leaves certain limitations in terms of rapid, high-throughput, and reproducible hypothesis testing. This is because primary cells are generally difficult and time-consuming to isolate, are limited in number, invariably contain impurities with other cell types, and may lack the features of pathological vasculature. Further, the isolation procedures themselves may affect cell viability and phenotypic homogeneity. Other disadvantages of primary cells include higher rates of bacterial or fungal contamination, a finite lifespan in cultures, and low-transfection efficiency. Although liver endothelial cell lines have been used by other groups,14, 15, 16 an immortalized and fully characterized cell line derived from murine HSECs is lacking.
Therefore, we have generated transformed sinusoidal endothelial cells (TSECs), an immortalized cell line derived from murine HSECs that have maintained endothelial characteristics and some HSEC-specific features despite serial passages. Cells have a typical endothelial morphology and limited fenestrations, and express numerous endothelial cell-specific markers. Functionally, TSECs migrate in response to angiogenic growth factors, form vascular tube-like structures on Matrigel, endocytose acetylated low-density lipoprotein (AcLDL), and secrete proteins involved in matrix remodeling. Overall, their characteristics and behavior most closely recapitulate liver endothelial cells that have undergone an angiogenic transformation, similar to the neovasculature associated with chronic liver disease. Importantly, the cells can be easily transduced with high efficiency using viral vectors. Collectively, therefore, the results of this study report the generation of TSECs, a cell line that should provide a homogeneous and unlimited culture model suitable for studying a broad range of liver endothelial cell biology, including motility and angiogenesis, and potentially more generalized endothelial cell biology as well.
MATERIALS AND METHODS
Isolation of Mouse HSECs
Freshly isolated mouse HSECs (mHSECs) were generated from whole mouse liver by mechanical disruption, enzymatic digestion, and immunomagnetic bead separation, as previously described, with modifications.13, 17, 18, 19 Briefly, liver tissue was harvested, dissected, washed, minced, digested in a collagenase buffer, and incubated with immunomagnetic Dynabeads (Dynal) coated with rat anti-mouse CD31 (BD Biosciences), an endothelial marker,19, 20, 21 for 1 h at room temperature. Cells were separated with a magnet and were plated on collagen-coated dishes. Viability was >90% by trypan blue staining and purity was >95% by staining for CD31.
Cell Culture
mHSECs or TSECs were grown in standard tissue culture conditions in endothelial cell media (ECM; ScienCell) containing 5% fetal bovine serum, 1% penicillin/streptomycin, and 1% ECGS (ScienCell). Bovine aortic endothelial cells (BAECs) were grown in standard tissue culture conditions in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. mHSECs were grown on collagen-coated cell culture vessels. TSECs and BAECs were grown on plastic dishes without a collagen coating. Doubling time was measured using manual counting on a hemacytometer.
Immortalization
mHSECs were immortalized using a pantropic lentivirus to overexpress the SV40 large T-antigen. Briefly, viral supernatant was produced in 293T cells and supernatant containing high-titer SV40 virus was diluted 1:2 in culture media and added to mHSECs 24 h after plating. Cells were incubated for 48 h, and then washed and cultured in ECM for 24 h. This process was repeated for a total of five cycles of transduction. SV40 expression was assessed using standard reverse transcription PCR (RT-PCR), as described below.
Cell Proliferation Assay
The cell proliferation rate of both mHSECs and TSECs was measured in 96-well plates using the non-radioactive cell proliferation assay (Promega). Optical density at 490 nm was measured with a plate reader at baseline and 48 h to calculate the proliferation rate.
Subcloning and Endocytosis of AcLDL
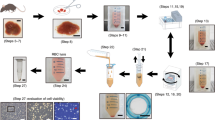
After immortalization, cells were split thinly into 96-well culture plates at an average density of 1 cell/well. After 48 h of expansion, the cells were trypsinized and passed to a 60 mm culture dish. The following day, cells were incubated with 10 μg/ml DiI-labled AcLDL (DiI-AcLDL; Invitrogen) for 2 h, washed, and imaged using confocal microscopy. Clones that had uniformly endocytosed DiI-AcLDL were further subcloned using cloning cylinders and were expanded in cultures. Figure 1 summarizes the isolation, immortalization, and subcloning techniques. Subsequent characterization was performed at passage numbers four to six after immortalization. Some studies were later repeated at passage number 20.

Isolation and immortalization scheme. HSECs were isolated from mouse liver by mechanical disruption, enzymatic digestion, and CD31-based immunomagnetic separation. HSECs were immortalized with SV40 large T-antigen and subcloned on the basis of their ability to endocytose AcLDL.
Light Microscopy
Standard light microscopy was performed at × 10 or × 20 using an Axiovert 40 CFL inverted microscope (Zeiss) and imaged with a ProgRes C3 digital camera system.
Electron Microscopy
Scanning electron microscopy was performed as previously described.22 mHSECs, TSECs, or BAECs were fixed in 2.5% glutaraldehyde for 1 h, post-fixed in 1% osmium tetroxide on ice for 30 min, dehydrated, critical-point dried, sputter or carbon coated, and imaged at 3 kV using an S-4700 scanning electron microscope (Hitachi).
RT-PCR
RNA from mHSECs or TSECs was isolated using the QiaShredder and RNeasy kits (Qiagen), according to the manufacturer's instructions. RNA was used for reverse transcription using the RT2 kit (SA Biosciences). Standard RT-PCR or cyber green-based real-time quantitative RT-PCR was performed using Taq polymerase (Invitrogen) or the Endothelial Cell Biology Array (SA Biosciences), respectively, according to the manufacturers’ instructions. Array data were processed using the PCR Array Data Analysis Web Portal (SA Biosciences).
Bioinformatics
Further rational processing of array data included ontological identification and pathway reconstruction. Ontological description of genes was provided by SA Biosciences. Pathway reconstruction was performed by processing the data using a semantic-based algorithm, as previously described.23 Data for this reconstruction were derived from a variety of databases, including Unigene, OMIN, Expasy, DIP (protein interactome), Biocarta, and Oncomime. Data were processed and manually curated using a computer interface provided by Ariadne Genomics, Inc.
Immunoblotting
Western blotting was performed as previously described.24 Briefly, mHSECs or TSECs was homogenized in lysis buffer and was cleared. In some experiments, serum-free cell culture supernatant was collected after treatment with 30 ng/ml vascular endothelial growth factor (VEGF), 25 ng/ml fibroblast growth factor (FGF), or vehicle for 24 h to blot for secreted proteins. 50 μg of each sample was denatured, electrophoresed, transferred, blocked, and incubated with antibodies to vWF (1:1000; Sigma), CD31 (1:1000; Santa Cruz), caveolin-1 (1:1000; BD Biosciences), fibronectin1 (1:1000; BD Biosciences), or actin (1:10 000; Sigma) for 1 h at room temperature. Horseradish peroxidase-conjugated secondary antibodies (GE healthcare) were used at 1:5000. Protein was detected using chemiluminescence (Santa Cruz) and autoradiography (Kodak).
Immunofluorescence
IF was performed as previously described.24 Approximately 20 000 cells were grown in 4-well chamber slides, fixed, quenched, blocked, and incubated with antibodies to vWF (1:250; Sigma), CD-31 (1:250; Santa Cruz), or caveolin-1 (1:250; BD Biosciences) overnight at 4°C. Fluorescent-tagged secondary antibodies were used at 1:500. Counterstaining was performed using TOTO-3. Cells were mounted and imaged using confocal microscope (Zeiss).
Chemotaxis Assays
Chemotaxis was measured by using a modified Boyden chamber assay (Becton Dickinson). Semi-permeable membranes were inserted into the chamber and mHSECs, TSECs, or BAECs were suspended in the serum-free medium in upper wells (20 000 cells/well) whereas lower chambers were filled with serum-free medium, 30 ng/ml VEGF, or 25 ng/ml FGF. After 6 h of incubation at 37°C, the membrane was removed and migrated cells were stained with DAPI. Random fields were imaged using fluorescence microscope and migrated cells were quantified in an automated manner using Metamorph software.
Vascular Tube Formation Assays
Vascular tube formation assays were performed on Matrigel or reduced growth factor Matrigel (BD Biosciences). Briefly, 4-well chamber slides (Lab-Tek) were coated with Matrigel or reduced growth factor Matrigel (150 μl/well), and mHSECs, TSECs, or BAECs were trypsinized and seeded onto the Matrigel (20 000 cells/well in 1 ml serum-free medium) in the presence or absence of 30 ng/ml VEGF or 25 ng/ml FGF. Random fields were photographed at 16 h after seeding. Vascular tube formation was assessed with an automated analysis of tube area using Metamorph software.
Transfection
Standard lipid-based transfection of plasmid DNA was performed using Effectene reagent (Qiagen) according to the manufacturer's specifications.
Viral Transduction
Adenoviruses were amplified and purified by the Gene Transfer Vector Core (University of Iowa). For adenoviral transduction, cells were washed and incubated for 1 h with 25 MOI of adenovirus encoding green fluorescent protein (GFP). Retroviral overexpression was performed using the pMMP-GFP retrovirus. Briefly, 293T cells were co-transfected with pMD.MLV gag.pol, pMD.G, and pMMP-GFP, using Effectene (Qiagen). The supernatant containing retrovirus was collected, diluted at 1:2, and added to mHSECs or TSECs.
Gelatin Zymography
mHSECs, TSECs, or BAECs were serum starved and treated with 30 ng/ml VEGF, 25 ng/ml FGF, or vehicle for 24 h. Cell culture supernatant was separated using SDS-PAGE containing 1 mg/ml gelatin. The gel was renatured for 30 min in 2.5% Triton X-100 and subsequently incubated for 24 h at 37°C in substrate buffer (50 mmol/l Tris-HCl, pH 7.5, containing 5 mmol/l CaCl2, 0.02% Brij-35) for matrix metalloproteinase (MMP) degradation of gelatin. Gels were stained with 0.5% Coomassie blue.
Statistical Analysis
Data are presented as mean±s.e.m. Bar graphs, blots, and micrographs represent typical experiments reproduced at least three times. Data analysis was performed using Graph Stat Prizm software. Data were analyzed for normal Gaussian distribution using the Kolmogorov–Smirnov normality test. For paired and normally distributed data, statistical analyses were performed using two-tailed Student's t-tests. For paired and non-normally distributed data, statistical analyses were performed using the Mann–Whitney U-test. For normally distributed multiple comparisons, statistical analyses were performed using one-way analysis of variance with a Tukey post hoc test. For all analyses, a P-value of <0.05 was considered statistically significant.
RESULTS
Isolation, Immortalization, and Subcloning
TSECs were generated as described in the methods section using CD31-based immunomagnetic separation, SV40 immortalization, and subcloning on the basis of the ability to endocytose AcLDL (Figure 1).
SV40 Expression
Using standard RT-PCR, we assessed the expression of the SV40 large T-antigen in both mHSECs and TSECs. As expected, TSECs showed robust expression, whereas mHSECs lacked expression (Figure 2a), indicating effective SV40 immortalization of TSECs.

Culture characteristics and morphology. (a) Standard RT-PCR shows expression of the SV40 large T-antigen in TSECs, but not in mHSECs. (b) Proliferation rate, measured over 48 h using a non-radioactive assay, shows the rapid growth characteristics of the TSEC cell line. (c) High-power phase contrast microscopic images of individual cells show formation of lamellipodia and filopodia in mHSECs, TSECs, and BAECs ( × 63). (d) Lower power images at confluence show a classic ‘cobblestone’ morphology of TSECs, similar to both mHSECs and BAECs ( × 20). *P<0.05.
Culture Characteristics and Morphology
After immortalization and subcloning, we directly compared the characteristics of mHSECs with the TSEC cell line. We found that whereas mHSECs required a collagen-coated culture surface, TSECs grew remarkably well on standard plastic cell culture vessels. TSECs grew extraordinarily fast with a doubling time of approximately 10 h and the proliferation rate 2.8-fold greater than that of mHSECs (Figure 2b), thereby increasing cell availability. Furthermore, many functional assays require long durations of serum starvation for adequate effect size and we found that whereas mHSECs revealed signs of toxicity and cell death after 24 h of serum starvation, TSECs tolerated serum starvation for periods as long as 96 h. Together, these results suggest that TSECs exist in an ‘activated’ state with increased adhesion, proliferation, and survival. By analyzing cells at low-density using phase contrast microscope, we found that TSECs formed lamellipodia and filopodia that were similar to those seen in both mHSECs and BAECs (Figure 2c). At confluence, TSECs also developed a classic ‘cobblestone’ morphology, typical of endothelial cells in cultures, but as expected, the primary mHSECs showed more heterogeneity, likely owing to small numbers of contaminating cell types, as compared with the more homogeneous TSEC morphology (Figure 2d).
Fenestrations
Transcytoplasmic fenestrations are small holes of ∼100–150 nm within the plasma membrane. The presence of these structures, organized into sieve plates, is one of the hallmark features, which is used to identify HSECs and it defines them as a specialized liver-specific endothelial cell.25 HSECs typically undergo defenestration in disease states and very quickly in cultures.26 Using scanning electron microscope at × 3500 and × 30 000, we compared the fenestrations of mHSECs with that of TSECs and also imaged BAECs as a negative control. We found that mHSECs maintained numerous transcytoplasmic fenestrations, similar to other isolated HSECs27 (Figures 3a and b; arrows). In contrast, BAECs, derived from aorta, lacked transcytoplasmic fenestrations (Figures 3c and d). Whereas many HSECs in long-term cultures lack fenestrations,28, 29 TSECs maintained a limited number of fenestrations between 100 and 150 nm that remained organized in sieve plates (Figures 3e–h; arrows). This markedly defenestrated state is most typical of a partially ‘capillarized’ HSEC phenotype, but the residual fenestrae confirm that the cells indeed have an HSEC origin. The presence of fenestrations in TSECs, along with their capacity for endocytosis, highlight some of the unique phenotypic characteristics of TSECs that differ from endothelial cell lines derived from other tissues.

TSECs maintain limited fenestrae organized in sieve plates. (a, b) Scanning electron microscopy with sputter coating shows that mHSECs visualized at (a) × 3500 or (b) × 30 000 have numerous fenestrae (arrows). (c, d) Scanning electron microscopy shows that BAECs visualized at (c) × 3500 or (d) × 30 000 lack fenestrae. (e–h) Scanning electron microscopy shows that TSECs maintain a limited number of fenestrae between 100–150 nm, organized in sieve plates (arrows). Panels represent: (e) sputter coating at × 3500, (f) sputter coating at × 30 000, (g) carbon coating at × 20 000, and (h) carbon coating at × 50 000.
TSECs Express Endothelial- and HSEC-Specific Markers
To confirm a broad endothelial phenotype, we analyzed TSECs by quantitative RT-PCR using the Endothelial Cell Biology Pathway Specific Quantitative PCR Array (SA Biosystems) and compared the genetic profile with that of mHSECs. Using this system, we found that TSECs maintained the expression of numerous endothelial cell-specific markers, including genes involved in vascular tone, angiogenesis, adhesion, extracellular matrix modulation, and thrombosis (Supplementary Table 1). In fact, the vast majority of genes on the Endothelial Cell Biology Array were expressed and most were detected at low-threshold cycle numbers, suggesting high levels of expression (Supplementary Figure 1). Comparing the mRNA expression profiles of mHSECs and TSECs, we found many genes expressed at similar levels (Figures 4a and b; black). A few genes were over-represented in TSECs, including Endothelin-2, Fibronectin1, MMP2, Integrin alpha V, and Serpine1 (Figures 4a and b; red). However, as might be expected in a cell culture model, we found that many genes were also downregulated after serial passage in cultures (Figures 4a and b; green). These changes in expression levels may be because of multiple factors, including SV40 expression, or loss of extracellular and paracrine cues from the in vivo microenvironment. We further analyzed the array data in silico with pathway reconstruction using a semantic-based algorithm. This showed that several important angiogenic pathways are highly represented in TSECs (Supplementary Figure 2). This information suggested that these cells may serve as a good model to study angiogenesis and, perhaps, broader endothelial cell biology as well. However, overall PCR data needs to be interpreted with caution as mRNA expression levels do not always correlate with protein levels. Using western blotting, we confirmed the presence of several liver endothelial markers in TSECs, including vWF, CD-31, and caveolin-1 (Figure 5a). IF staining showed these proteins to be present in all cells with a cytoplasmic and plasma membrane distribution (Figure 5b). Although both vWF25, 30, 31, 32, 33, 34 and CD3119, 20, 21 are widely used to identify liver endothelial cells, the ideal markers to identify these cells remains controversial27 and some studies suggest that CD31 should be regarded as more typical of the non-fenestrated cells seen after capillarization.28

TSECs maintain an endothelial genetic signature. (a) TSECs maintain an mRNA expression profile similar to mHSECs, as assessed by real time quantitative RT-PCR using the SA Biosciences Endothelial Cell Biology Pathway Specific Quantitative PCR Array. Scatter plot shows the number of genes with similar expression levels (within black lines), upregulated (red), or downregulated (green) (b). Heat mapping represents the fold-change in expression in TSECs, as compared with mHSECs. See also Supplementary Figures 1, 2, and 3 for gene names with fold change values, CT ranges, and pathway analysis, respectively.

TSECs maintain HSEC-specific markers and endocytic capacity. (a) Representative immunoblots (50 μg per lane) show that TSECs maintain expression of vWF, CD31, and caveolin-1, as compared with mHSECs. (b) IF staining confirms homogeneous expression and shows a cytoplasmic and plasma membrane distribution of vWF, CD31, and caveolin-1 (red) with nuclear counterstaining (blue). (c). Endocytosis assays were performed as described and imaged using fluorescence confocal microscope to show that TSECs maintain the ability to endocytose DiI-labeled AcLDL particles, as compared with mHSECs.
Endocytosis of AcLDL Particles
Endocytosis is a key function of normal HSEC physiology and this feature is widely exploited to identify HSECs in cultures.27 Indeed, we used the ability of cells to take up DiI-labled AcLDL during the initial subcloning of TSECs to select a pure clone. To confirm that TSECs maintain the ability to endocytose AcLDL, we preformed uptake studies in both mHSECs and in TSECs using DiI-labled AcLDL particles. Our studies show that both mHSECs and TSECs maintain the ability to endocytose the fluorescent-tagged LDL particles (Figure 5c). We did encounter reduced uptake of AcLDL in TSECs at passage number 20 (data not shown), indicating that endocytosis studies may be best performed at earlier passage numbers.
Chemotaxis in Response to Angiogenic Stimuli
Endothelial cell chemotaxis is required for angiogenesis and our pathway reconstruction suggested that angiogenic pathways are highly represented in TSECs. We therefore subjected mHSECs, TSECs, and BAECs to chemotaxis assays in response to multiple angiogenic growth factor stimuli using a modified Boyden Chamber. We found that TSECs had a moderate basal migration rate through a semi-permeable membrane and a chemotactic response to both VEGF and FGF (Figure 6a). When we quantified the migration data from the three cell types, we found that compared with mHSECs, TSECs showed a similar pattern of basal migration and 1.7- or 2.6-fold increases in response to VEGF and FGF, respectively, responses that were similar to the well-studied non-sinusoidal endothelial cell line, BAECs (Figure 6b). The above chemotaxis studies were performed in TSECs at passage number twenty indicating that motility responses are stable in TSECs, even at high passage numbers.

Chemotaxis in response to angiogenic growth factors. (a) TSECs were subjected to chemotaxis assays in the presence or absence of 30 ng/ml VEGF or 25 ng/ml FGF. Migrated cells are stained with DAPI. (b) mHSECs, BAECs, and TSECs were subjected to chemotaxis assays in the presence or absence of 30 ng/ml VEGF or 25 ng/ml FGF. Migrated cells are stained with DAPI (blue) and quantified using Metamorph software (n=7; mean±s.e.). *P<0.05 versus basal by Student's t-test. #P<0.05 versus basal by Mann–Whitney U-test.
Vascular Tube Formation
To further assess the angiogenic phenotype of TSECs, we subjected mHSECs, TSECs, and BAECs to vascular tube formation assays, a commonly used angiogenesis assay, on regular or growth factor reduced Matrigel, in the presence or absence of VEGF or FGF. We found that mHSECs showed negligible ability to form vascular tubes either in the basal state or in response to angiogenic factors (data not shown). TSECs, in contrast, formed robust vascular tube-like structures on reduced growth factor Matrigel alone and showed 3.3- or 2.4-fold increases in response to VEGF and FGF, respectively (Figure 7a), indicating an enhanced angiogenic phenotype. We noticed reductions in tube forming ability at passage number 20 (data not shown), indicating that tubulogenesis may be best studied at passage numbers earlier than this. On regular Matrigel (containing multiple growth factors), TSECs showed superior tube forming ability as compared with the widely studied BAECs line (Figure 7b).

Vascular tube formation on Matrigel. (a) TSECs were subjected to vascular tube formation assays on reduced growth factor Matrigel in the presence or absence of 30 ng/ml VEGF or 25 ng/ml FGF. Vascular tube structures were imaged using standard light microscope and quantified using Metamorph software (n=5; mean±s.e.). (b) BAECs and TSECs were subjected to vascular tube formation assays on complete Matrigel. Vascular tube structures were imaged using standard light microscope and quantified using Metamorph software (n=5; mean±s.e.). #P<0.05 versus basal using analysis of variance with Tukey post test. *P<0.05 versus BAECs using Student's t-test.
Extracellular Matrix Modulation
Our PCR array data suggested that some matrix modification proteins are highly expressed in TSECs, including fibronectin1 and MMP2, whereas others, such as MMP9, had lower expression levels. As altered potential for matrix remodeling may be important in facilitating angiogenic invasion during cirrhosis, we sought to confirm that TSECs indeed have enhanced potential for modification of extracellular matrix. We treated mHSECs, TSECs, and BAECs with VEGF, FGF, or vehicle and assayed for the secretion of fibronectin1, MMP2, and MMP9. Using western blotting, we found that basal secretion of fibronectin1 was higher in TSECs than in mHSECs, although incremental increases in secretion were not evident after treatment with angiogenic factors. Using gelatin zymography, we found that mHSECs showed inducible secretion of both MMP2 and MMP9. TSECs, in contrast, had constitutively high activity of MMP2, but reduced activity of MMP9, consistent with the expression changes observed by real-time PCR array. These data are broadly consistent with an activated phenotype in TSECs, including enhanced matrix turnover, a characteristic that may allow for more efficient angiogenic invasion through the fibrotic microenvironment in the setting of cirrhosis and portal hypertension.
Transfection and Transduction Efficiency
In vitro experiments seeking mechanistic insight frequently require the ability to genetically modify cultured cells in an efficient manner. Primary cells are notoriously difficult to transfect and this technical obstacle can, at times, limit opportunities for molecular and genetic interventions. We therefore performed a series of plasmid transfection and viral transduction experiments using both mHSECs and TSECs. We found that standard transfection of plasmid DNA for GFP was virtually impossible in mHSECs, with negligible transfection rates and cellular toxicity (data not shown). TSECs also showed low-transfection efficiency of <5% by standard lipid-based transfection methods (Figure 8). Using an adenoviral approach, we were able to improve the efficiency in both cell types, with TSECs achieving approximately 40% transduction efficiency at 25 MOI (Figure 8), a number that could likely be increased further with higher viral titers. In addition, whereas retroviral transduction was unsuccessful in mHSECs, perhaps because of their low-proliferation rate, this approach achieved approximately 90% transduction efficiency in TSECs (Figure 8). In addition to high transduction efficiency and robust overexpression, retroviral systems in TSECs have the added advantage of providing stable overexpression, eliminating the need to transfect repeatedly for each individual experiment.

Transfection and transduction efficiency. (a–c) Representative overlay images of phase contrast and fluorescence microscopy show the transfection and transduction efficiency of TSECs for GFP using lipid-based transfection (a), adenoviral transduction (b), or retroviral transduction (c). (d) Quantification of transfection and transduction efficiency in TSECs for GFP using lipid-based transfection, adenoviral transduction, or retroviral transduction (n=6; mean±s.e.).
DISCUSSION
In the current study, we describe the development of TSECs, a new cell line derived from murine HSECs. We characterize both a broad endothelial phenotype as well as some HSEC-specific features of these cells using molecular, biochemical, morphologic, ultrastructural, and functional approaches. Overall, the TSECs phenotype is most similar to an ‘activated’ form of liver endothelia in which cells become proliferative, defenestrated, and angiogenic, as seen in states of chronic liver disease. However, the cells also retain characteristics unique to HSECs, including transcytoplasmic fenestrations, endocytic capacity, and expression of specific protein markers. Direct comparisons with both mHSECs and BAECs point out the similarities, differences, and some technical advantages of TSECs.
TSECs are immortalized with SV40 large T-antigen and have been stable in cultures for over 9 months and 30 passages, suggesting that these cells will be a reliable and reproducible long-term cell culture model. However, we also noticed some phenotypic changes occurring in cells above passage number twenty (specifically, reduced endocytosis and tubulogenesis), indicating that some studies may be best performed at lower passage numbers. This still represents a significant advantage over primary cells, which are generally not passed in cultures and over BAECs, which can only be used at very low-passage numbers. Other TSECs characteristics, such as chemotaxis, actually remained quite stable after multiple passages. We show that the light microscopic features of TSECs are characteristic of endothelial cells in cultures and parallel the findings in both mHSECs and BAECs. Their rapid proliferation, tolerance of serum starvation, and ability to grow on uncoated dishes offer technical advantages over primary cells. The presence of a limited number of fenestrae organized in sieve plates is a reassuring sign of an HSEC origin and is unique compared with cell lines from non-liver sources.25 However, the relative defenestration compared with mHSECs also suggests some dedifferentiation toward a cell type more reminiscent of pathologic vasculature.
Our PCR array data show that TSECs broadly retain an endothelial genetic signature, expressing numerous endothelial genes, including many involved in vascular tone, angiogenesis, adhesion, modulation of extracellular matrix, and thrombosis. Furthermore, their overall genetic profile is similar to mHSECs and bioinformatics approaches suggest that angiogenic pathways are highly represented in TSECs. Liver endothelia have historically been identified by the expression of specific marker genes, such as vWF25, 30, 31, 32, 33, 34 and CD31.19, 20, 21 Recent studies, however, point out considerable heterogeneity both within and among the species, as well as plasticity of expression patterns depending on disease states, differences between cells in vivo and cultured cells, and variability depending on the length of time in cultures.26, 27, 28 Recognizing these discrepant reports, we, nevertheless, are able to show that TSECs maintain protein expression of some of the most well-characterized markers of liver endothelial cells, including vWF and CD31.
TSECs maintain many important functional characteristics of endothelial cells in general and of HSECs in particular. They migrate in response to the angiogenic growth factors VEGF and FGF, form vascular tubes on Matrigel, endocytose AcLDL, and secrete proteins involved in matrix remodeling. The functional angiogenic response of TSECs, in terms of tubulogenesis and invasion potential, seems to be greatly enhanced relative to primary cells, highlighting some inherent limitations of primary cell isolation preps and/or indicating dedifferentiation of TSECs toward an angiogenic phenotype. Regardless, the larger and more consistent effect sizes seen in TSECs offers a technical advantage in performing pro- and anti-angiogenic studies. The limited ability of mHSECs to form tubes effectively is a common problem with primary isolates and is likely because of some degree of trauma/toxicity from the isolation procedure itself. TSECs overcome these issues by providing a cell type with robust angiogenic responses and consistency of results.
Although immortalized cell lines in cultures inherently differ from their in vivo counterparts owing to dedifferentiation in cultures, changes related to immortalization itself, and the loss of paracrine and other microenvironmental cues, they can provide powerful in vitro tools for rapid screening of hypotheses and molecular interventions needed for deeper mechanistic insights. Immortalized cell lines can overcome many of the disadvantages of primary cells by providing a robust, reproducible, and unlimited model for advancing a field quickly. Indeed, examples in other liver disciplines, including fibrogenesis35 and cholangiocyte biology,36, 37 have revealed that periods of significant scientific expansion in our understanding of a field can be facilitated by development of appropriate experimental models, including immortalized cell culture lines.
Although several endothelial cell lines from non-liver sources exist, a model system for studying the unique structure and function of endothelial cells derived from the hepatic sinusoid, as well as the pathological changes they undergo during chronic liver disease was lacking. TSECs maintain not only the three key features of these cells (fenestrations, endocytic capacity, and specific protein markers), but also recapitulate many of the changes seen in chronic liver disease. TSECs are likely useful for the study of a variety of normal and pathological functions of liver endothelial cells, but they may be particularly well suited for studies on cell motility, matrix invasion, and angiogenesis. Given the relative rarity of endothelial cell lines in general, TSECs may also be of broader interest to those studying angiogenesis and endothelial cell biology outside the liver.
References
Shah V . Hepatic circulation. Encyclopedia of Gastroenterology. Elsevier: USA, 2004, pp 299–304.
Huebert RC, Shah V . Hepatic sinusoidal endothelial cells. In: Dufour JF, Clavien PA (eds). Signaling Pathways in Liver Diseases. Springer-Verlag: Berlin Heidelberg, 2009, pp 79–91 (in press).
Vanheule E, Geerts AM, Van Huysse J, et al. An intravital microscopic study of the hepatic microcirculation in cirrhotic mice models: relationship between fibrosis and angiogenesis. Int J Exp Pathol 2008;89:419–432.
Tugues S, Fernandez-Varo G, Munoz-Luque J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology 2007;46:1919–1926.
Fernandez M, Semela D, Bruix J, et al. Angiogenesis in liver disease. J Hepatol 2009;50:604–620.
Lalor PF, Lai WK, Curbishley SM, et al. Human hepatic sinusoidal endothelial cells can be distinguished by expression of phenotypic markers related to their specialised functions in vivo. World J Gastroenterol 2006;12:5429–5439.
Iwakiri Y, Groszmann RJ . Vascular endothelial dysfunction in cirrhosis. J Hepatol 2007;46:927–934.
Shah V . Cellular and molecular basis of portal hypertension. Clin Liver Dis: Portal Hypertens 2001;5:629–644.
Yang ZF, Poon RT . Vascular changes in hepatocellular carcinoma. Anat Rec (Hoboken) 2008;291:721–734.
O’Reilly JN, Cogger VC, Le Couteur DG . Old age is associated with ultrastructural changes in isolated rat liver sinusoidal endothelial cells. J Electron Microsc (Tokyo) 2010;59:65–69.
Knook DL, Sleyster EC . Separation of Kupffer and endothelial cells of the rat liver by centrifugal elutriation. Exp Cell Res 1976;99:444–449.
Tokairin T, Nishikawa Y, Doi Y, et al. A highly specific isolation of rat sinusoidal endothelial cells by the immunomagnetic bead method using SE-1 monoclonal antibody. J Hepatol 2002;36:725–733.
LeCouter J, Moritz DR, Li B, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science 2003;299:890–893.
Matsuura T, Kawada M, Hasumura S, et al. High density culture of immortalized liver endothelial cells in the radial-flow bioreactor in the development of an artificial liver. Int J Artif Organs 1998;21:229–234.
Henning W, Bohn W, Nebe B, et al. Local increase of beta 1-integrin expression in cocultures of immortalized hepatocytes and sinusoidal endothelial cells. Eur J Cell Biol 1994;65:189–199.
Matsumura T, Takesue M, Westerman KA, et al. Establishment of an immortalized human-liver endothelial cell line with SV40T and hTERT. Transplantation 2004;77:1357–1365.
Zirlik A, Bavendiek U, Libby P, et al. TRAF-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Arterioscler Thromb Vasc Biol 2007;27:1101–1107.
Lalor PF, Edwards S, McNab G, et al. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J Immunol 2002;169:983–992.
Follenzi A, Benten D, Novikoff P, et al. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest 2008;118:935–945.
Zhou W, Inada M, Lee TP, et al. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest 2005;85:780–788.
Benten D, Follenzi A, Bhargava KK, et al. Hepatic targeting of transplanted liver sinusoidal endothelial cells in intact mice. Hepatology 2005;42:140–148.
Masyuk AI, Gradilone SA, Banales JM, et al. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am J Physiol Gastrointest Liver Physiol 2008;295:G725–G734.
Nikitin A, Egorov S, Daraselia N, et al. Pathway studio—the analysis and navigation of molecular networks. Bioinformatics 2003;19:2155–2157.
Huebert RC, Splinter PL, Garcia F, et al. Expression and localization of aquaporin water channels in rat hepatocytes. Evidence for a role in canalicular bile secretion. J Biol Chem 2002;277:22710–22717.
Shah V, Haddad F, Garcia-Cardena G, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of hepatic resistance. J Clin Invest 1997;100:2923–2930.
Smedsrod B, Le Couteur D, Ikejima K, et al. Hepatic sinusoidal cells in health and disease: update from the 14th International Symposium. Liver Int 2009;29:490–501.
Elvevold K, Smedsrod B, Martinez I . The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol 2008;294:G391–G400.
DeLeve LD, Wang X, McCuskey MK, et al. Rat liver endothelial cells isolated by anti-CD31 immunomagnetic separation lack fenestrae and sieve plates. Am J Physiol Gastrointest Liver Physiol 2006;291:G1187–G1189.
March S, Hui EE, Underhill GH, et al. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology 2009;50:920–928.
Eyhorn S, Schlayer HJ, Henninger HP, et al. Rat hepatic sinusoidal endothelial cells in monolayer culture. Biochemical and ultrastructural characteristics. J Hepatol 1988;6:23–35.
Shaw RG, Johnson AR, Schulz WW, et al. Sinusoidal endothelial cells from normal guinea pig liver: isolation, culture and characterization. Hepatology 1984;4:591–602.
Rieder H, Ramadori G, Dienes HP, et al. Sinusoidal endothelial cells from guinea pig liver synthesize and secrete cellular fibronectin in vitro. Hepatology 1987;7:856–864.
Soda R, Tavassoli M . Insulin uptake by rat liver endothelium studied in fractionated liver cell suspensions. Mol Cell Biochem 1985;65:117–123.
Gatmaitan Z, Varticovski L, Ling L, et al. Studies on fenestral contraction in rat liver endothelial cells in culture. Am J Pathol 1996;148:2027–2041.
Xu L, Hui A, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 2005;54:142–151.
Vroman B, LaRusso NF . Development and characterization of polarized primary cultures of rat intrahepatic bile duct epithelial cells. Lab Invest 1996;74:303–313.
Muff MA, Masyuk TV, Stroope AJ, et al. Development and characterization of a cholangiocyte cell line from the PCK rat, an animal model of autosomal recessive polycystic kidney disease. Lab Invest 2006;86:940–950.
Acknowledgements
We acknowledge Helen Hendrickson for technical support, Angela Mathison for assistance with pathway analysis, and Theresa Johnson for secretarial support. This work was supported by grants DK59615-06 (Shah), HL086990 (Shah), DK24031 (LaRusso), P30DK084567 (LaRusso), and the Loan Repayment Program (Huebert) from the National Institutes of Health; by the Hartz Foundation; and the Mayo Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Rights and permissions
About this article
Cite this article
Huebert, R., Jagavelu, K., Liebl, A. et al. Immortalized liver endothelial cells: a cell culture model for studies of motility and angiogenesis. Lab Invest 90, 1770–1781 (2010). https://doi.org/10.1038/labinvest.2010.132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.132
Keywords
This article is cited by
-
Treprostinil reduces endothelial damage in murine sinusoidal obstruction syndrome
Journal of Molecular Medicine (2019)
-
A Pathway to Personalizing Therapy for Metastases Using Liver-on-a-Chip Platforms
Stem Cell Reviews and Reports (2017)
-
Development and characterization of human-induced pluripotent stem cell-derived cholangiocytes
Laboratory Investigation (2015)

{kind=link}
{kind=link}
{kind=link}
{kind=link}