
Abstract
Understanding the chemistry of protein modification by formaldehyde fixation and subsequent tissue processing is central to developing improved methods for antigen retrieval in immunohistochemistry and for recovering proteins from formalin-fixed, paraffin-embedded (FFPE) tissues for proteomic analysis. Our initial studies of single proteins, such as bovine pancreatic ribonuclease A (RNase A), in 10% buffered formalin solution revealed that upon removal of excess formaldehyde, monomeric RNase A exhibiting normal immunoreactivity could be recovered by heating at 60°C for 30 min at pH 4. We next studied tissue surrogates, which are gelatin-like plugs of fixed proteins that have sufficient physical integrity to be processed using normal tissue histology. Following histological processing, proteins could be extracted from the tissue surrogates by combining heat, detergent, and a protein denaturant. However, gel electrophoresis revealed that the surrogate extracts contained a mixture of monomeric and multimeric proteins. This suggested that during the subsequent steps of tissue processing protein–formaldehyde adducts undergo further modifications that are not observed in aqueous proteins. As a first step toward understanding these additional modifications we have performed a comparative evaluation of RNase A following fixation in buffered formaldehyde alone and after subsequent dehydration in 100% ethanol by combining gel electrophoresis, chemical modification, and circular dichroism spectroscopic studies. Our results reveal that ethanol-induced rearrangement of the conformation of fixed RNase A leads to protein aggregation through the formation of large geometrically compatible hydrophobic β-sheets that are likely stabilized by formaldehyde cross-links, hydrogen bonds, and van der Waals interactions. It requires substantial energy to reverse the formaldehyde cross-links within these sheets and regenerate protein monomers free of formaldehyde modifications. Accordingly, the ethanol-dehydration step in tissue histology may be important in confounding the successful recovery of proteins from FFPE tissues for immunohistochemical and proteomic analysis.
Similar content being viewed by others

Main
Formaldehyde fixation and paraffin embedding remains the standard technique for preserving tissue specimens for pathological examination and the study of tissue morphology. Archival repositories now contain millions of formalin-fixed, paraffin-embedded (FFPE) tissue samples, which provide an invaluable resource for the retrospective study of disease progression and response to therapy. In many cases, malignant cells yield unique ‘protein profiles’ when total protein extracts from such cells are analyzed by two-dimensional gel electrophoresis or mass spectrometry methods.1,2,3,4,5 Such proteomic studies of malignant tissues have the potential to provide an important complement to the genomic analysis of these tissues.6 A severe limitation of proteomic studies using fresh or frozen tissue is that the results cannot be related directly to the clinical course of diseases, particularly those where the time between treatment and recurrence is long. However, if routinely fixed and embedded archival tissues could be used for standard proteomic methods these powerful techniques could be used to both qualitatively and quantitatively analyze large numbers of tissues for which the clinical course of disease has been established.
Analysis of archival FFPE tissues by high-throughput proteomic methods has been hampered by the adverse effects of formaldehyde fixation.2 Formaldehyde–protein adducts, in addition to both intra- and intermolecular cross-links, are formed during tissue fixation.7,8 These formaldehyde-induced modifications reduce protein immunoreactivity and extraction efficiency, and they may lead to misidentification of proteins during proteomic analysis.9 Thus, understanding the chemistry of protein modification by formalin-fixation, dehydration, and paraffin embedding is central to developing improved methods for recovering protein from FFPE tissues for proteomic analysis and to improve antigen-retrieval methods for immunohistochemistry. Metz et al7,8 have identified three types of chemical modifications after treatment of proteins with formaldehyde: (1) methylol (hydroxymethyl) adducts, (2) Schiff bases, and (3) stable methylene bridges. Formaldehyde can react with lysine, cysteine, arginine, tryptophan, histidine, and the N-terminal amine to form methylol adducts. The methylol adduct can subsequently undergo a dehydration reaction to form a Schiff base, which is seen most frequently in lysine and tryptophan residues. Both methylol and Schiff-base adducts were shown to form intermolecular cross-links. In addition, the protein N-terminal amine can be converted to a stable 4-imidazolidinone adduct.8 Intramolecular protein cross-links (methylene bridges) have been reported in both model peptides7 and whole proteins, such as insulin.8
Previously we have modeled the effects of formaldehyde fixation of proteins in solution. In these studies, incubation of 1–6.5 mg/ml solutions of ribonuclease A (RNase A) in 10% aqueous formalin leads to the formation of extensive intra- and intermolecular cross-links, as observed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Differential scanning calorimetry studies demonstrated that these cross-links increased the thermal denaturation temperature of RNase A, whereas optical spectroscopic studies indicated that formaldehyde fixation does not appear to significantly alter the protein's secondary or tertiary structure.10 Mild heating of the formaldehyde-treated RNase A solutions at 65°C in pH 4 buffer resulted in almost quantitative reversal of formaldehyde cross-links, which lead to the recovery of both immunoreactivity and enzyme function.11
These studies do not address, however, the effects of further processing on fixed proteins. In a previous study, we utilized one and two protein ‘tissue surrogates’ to model FFPE tissues.12 Cytoplasmic proteins, such as lysozyme or RNase A, at concentrations approaching the protein content in whole cells, were fixed with 10% formalin to form gelatin-like plugs. These plugs had sufficient physical integrity to be processed through graded alcohols, xylene, and embedded in paraffin according to standard histological procedures.13 Optimal protein extraction from these tissue surrogates was obtained by combining heat, a detergent, and a protein denaturant. However, SDS–PAGE analysis revealed that the reversal of the intermolecular cross-links required high temperatures (>100°C),12 or the application of elevated hydrostatic pressure at moderate temperatures.14 This suggests that in fixed, dehydrated, and paraffin-embedded tissues, protein–formaldehyde adducts undergo further reactions that are not observed in aqueous solution and require additional energy to reverse formaldehyde-induced modifications. As a first step toward understanding these additional modifications we report here a comparative evaluation of RNase A following fixation in buffered formaldehyde solution alone, and after subsequent dehydration in 100% ethanol.
MATERIALS AND METHODS
Bovine pancreatic RNase A (type III-A), pyridoxal 5′-phosphate (PLP), and sodium cyanoborohydride (NaBH3CN) were purchased from Sigma (St Louis, MO, USA). Aqueous formaldehyde (37% w/w) was purchased from Fisher Scientific (Fair Lawn, NJ, USA). Absolute ethanol was purchased from Pharmco-AAPER (Brookfield, IL, USA).
Formaldehyde Fixation and Ethanol Dehydration
Volumes of native RNase A at 4 mg/ml in 10 mM sodium phosphate buffer, pH 7.4, were treated for 24 h with an equal volume of 20% (v/v) phosphate-buffered formalin. The final concentrations were 2 mg/ml protein and 10% formalin in 10 mM sodium phosphate, pH 7.4. After incubating overnight, the formaldehyde-fixed proteins were divided into equal aliquots. The formaldehyde from half of the 24-h fixed samples was removed by washing the aliquot five times with deionized (DI) water in a Microcon YM-3 concentrator (Millipore, Billerica, MA, USA).
Potassium phosphate (50 mM, pH 7.0), was added to the remaining aliquots (8.3 mM, final concentration) and the protein was precipitated in 10 volumes of ice-cold acetone. The pellet was washed with 1 ml of ice-cold acetone and then dried under vacuum for 15 min. The precipitated, fixed protein was then incubated under 1 ml of 100% EtOH at room temperature. After 1 h, 24 h, or 1 week, the precipitated protein was pelleted by centrifugation, and the ethanol was removed. The air-dried pellets were then resuspended in 10 mM potassium phosphate buffer, pH 7.4, or in DI water to their original volumes.
Trapping of Formaldehyde Adducts with NaBH3CN or PLP
The final protein concentration of aliquots of 24-h formaldehyde-fixed RNase A (with the formaldehyde removed) or native RNase A was adjusted to 2 mg/ml in 20 mM phosphate buffer, pH 7.4. Equal volumes of NaBH3CN in DI water were added to 50 M excess and the pH of the samples was adjusted to 8.0 with 0.1 M NaOH, using a previously published protocol.15 After incubating at room temperature for 4 h, the excess NaBH3CN was quenched by adding 2.5% (v/v) of a 1 M HCl solution. Alternately, the native or formaldehyde-fixed RNase A was incubated in the presence of 0.1–10 mM PLP for 4 h at room temperature.16 The protein solutions were then precipitated in 10 volumes of ice-cold acetone as described earlier and incubated under 1 ml of 100% ethanol for up to 1 week. The NaBH3CN or PLP-treated RNAse A was resuspended in DI water to final concentration of 2 mg/ml prior to protein recovery.
Protein Recovery
All native, formaldehyde-fixed, and resuspended formaldehyde-fixed, ethanol-treated protein samples were added to an equal volume of a recovery buffer containing 4% SDS in 40 mM Tris-HCl, pH 4.0, for a final protein concentration of 1 mg/ml. The protein samples were then heated at 100°C for 20 min, followed by 60°C for 2 h, according to the antigen-retrieval protocol of Shi et al.17 After protein recovery, any remaining unsolubilized material was pelleted at 14 000 g for 20 min and the supernatant was saved for further analysis. The antigen-retrieved RNase A solutions were then diluted directly into sample buffer and analyzed by SDS–PAGE with no further heating.
Analysis of Protein Composition
After sample processing, 20 ml of each native, formaldehyde-fixed, and formaldehyde-fixed, ethanol-treated RNase A preparation was diluted directly into 4 × LDS sample buffer supplemented with 10 × sample-reducing buffer (Invitrogen, Carlsbad, CA, USA) with no additional heat treatment. SDS–PAGE was performed on precast NuPAGE Bis-Tris 4–12% gradient polyacrylamide gels (1 × 80 × 80 mm) using 2-(N-morpholino)ethanesulfonic acid–SDS running buffer at pH 7.3 (Invitrogen). Molecular mass standards and the Coomassie blue-based colloidal staining kit were also purchased from Invitrogen. Gel images were documented using a Scanmaker i900 (Microtek, Carson, CA, USA), and annotated in Adobe Photoshop. The composition of individual gel lanes was analyzed and percentages were determined using the Un-Scan-it Gel 6.1 program (Silk Scientific Corp., Orem, UT, USA).
Spectroscopic Measurements
Circular dichroism (CD) spectra in the near-UV region (246–350 nm) were acquired from native, formaldehyde-fixed, and formaldehyde-fixed RNase A incubated in ethanol for 1 week using a 10-mm path length water-jacketed cell. CD spectra in the far-UV region (180–250 nm) were recorded using a 0.02-mm path length water-jacketed cell. The CD spectra were recorded at 24°C with a Jasco-715 spectropolarimeter equipped with an external water bath (Jasco Corp., Easton, MD, USA). Each spectrum was an average of 10 measurements taken under identical conditions. The following settings were used: the scan speed was 50 nm/min, the bandwidth was 1 nm, the step resolution was 0.2 nm, and the response time was 8 s. The native, formaldehyde-fixed, and formaldehyde-fixed, ethanol-precipitated proteins were reconstituted in 10 mM phosphate buffer, pH 7.4, prior to analysis by CD spectropolarimetry. The ethanol-precipitated samples were only partially soluble in phosphate buffer. The formaldehyde-fixed RNase A was analyzed directly in 10% phosphate-buffered formaldehyde. The final protein concentration of each RNase sample was adjusted to 0.65 mg/ml, as determined spectrophotometrically assuming that the E1%=7.3 at 280 nm.18
RESULTS
Formaldehyde Fixation, Ethanol Treatment, and Protein Recovery
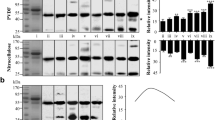
After fixation in 10% buffered formalin, gel electrophoresis of 1 mg/ml solutions of RNase A showed a mixture of intermolecular cross-linked proteins composed of monomeric (25%), dimeric (21%), trimeric (18%), tetrameric (15%), pentameric (10%), and hexameric (11%) species (Figure 1, lane 2). Removal of the formaldehyde did not reduce the level of cross-linking (data not shown), however, heating the formaldehyde-fixed sample in 20 mM Tris-HCl with 2% SDS at pH 4 resulted in an almost fourfold increase in monomeric protein, with approximately 8% of total protein composed of dimeric protein (Figure 1, lane 3). This is consistent with previously reported studies of fixed RNAse at concentrations of 1 mg/ml.10,11 To mimic the ethanol-dehydration step typically performed during the histological processing of FFPE tissues,19 the formaldehyde-fixed RNase A was precipitated and incubated in 100% ethanol for 1 h, 24 h, or 1 week. SDS–PAGE revealed that the formaldehyde-fixed, ethanol-treated samples were as highly cross-linked as the formaldehyde-fixed samples (Figure 1, lanes 4, 6, and 8). However, after heating in the Tris–SDS recovery buffer at 100°C for 20 min, followed by 60°C for 2 h, no reversal of the formaldehyde-induced cross-links was observed (Figure 1, lanes 5, 7, and 9), with the total protein content corresponding to 26% monomeric, 23% dimeric, 18% trimeric, 14% tetrameric, 11% pentameric, and 8% hexameric species.

SDS–PAGE of formaldehyde-fixed RNase A before and after protein retrieval. Lane M: molecular weight marker; lane 1: native RNase A; lane 2: formaldehyde-fixed RNase A after the removal of excess formaldehyde; lane 3: formaldehyde-fixed RNase A sample from lane 2 after retrieval in 20 mM Tris-HCl, pH 4.0, with 2% SDS; lanes 4, 6, and 8: formaldehyde-fixed RNase A after incubation in 100% ethanol for 1 h, 24 h, or 1 week, respectively; lanes 5, 7, and 9: 1 h, 24 h, or 1 week formaldehyde-fixed, ethanol-treated RNase A after retrieval in 20 mM Tris-HCl, pH 4.0, with 2% SDS. The RNase A samples were heated at 100°C for 20 min, followed by 60°C for 2 h.
Effect of PLP or NaBH3CN on Formaldehyde-Induced Cross-Links
To further examine the nature and mechanism of the formation of formaldehyde-induced cross-links in RNase A, we studied the effects of treatment with the reducing agent NaBH3CN or PLP. Fixed RNase A solutions (2 mg/ml) were dialyzed against 10 mM potassium phosphate to remove excess formaldehyde. The fixed protein solutions were then treated with 0.1–10 mM PLP, which has been shown to reversibly react with Schiff bases, but may also react with hydroxymethyl adducts and amine groups.16,20 After 4-h incubation at neutral pH, the RNase A solutions were precipitated to remove excess PLP and incubated under ethanol for 24 h. The small amount of protein dimer present in the control sample (native RNase treated with PLP and ethanol; Figure 2, lane 1) is attributable to an impurity in the RNase from Sigma (Figure 1, lane 1). In the absence of protein recovery, treatment with PLP did not prevent the formation of protein oligomers, with total protein content consisting of approximately 45% monomeric protein (Figure 2, lanes 2, 4, and 6). However, after heating in 20 mM Tris-HCl with 2% SDS at pH 4, there were greater degrees of recovery of monomeric protein with increasing concentrations of PLP (Figure 2, lanes 3, 5, and 7), with total protein content consisting of 70, 81, and 90% monomeric RNase A for the samples treated with 0.1, 1, and 10 mM PLP, respectively (Figure 2, lanes 3, 5, and 7).

The effect of PLP on the formation of intermolecular cross-links. Lane M: molecular weight marker; lane 1: native RNase A treated with PLP and incubated in 100% ethanol for 24 h; lanes 2, 4, and 6: formaldehyde-fixed RNase A treated with 0.1, 1, and 10 mM PLP and incubated under 100% ethanol for 24 h; lanes 3, 5, and 7: samples from lanes 2, 4, and 6, respectively, after protein retrieval in 20 mM Tris-HCl, pH 4.0, with 2% SDS. The RNase A samples were heated at 100°C for 20 min, followed by 60°C for 2 h.
We next treated the formaldehyde-fixed RNase A solutions with a 50 M excess of NaBH3CN, which has been shown to reduce a wide variety of organic functional groups, including Schiff bases and hydroxymethyl groups, at neutral pH.15,21,22 SDS–PAGE analysis revealed that formaldehyde-fixed RNase A reduced with NaBH3CN and then incubated in ethanol exhibited significantly fewer intermolecular cross-links (Figure 3, lane 2) than their nonreduced counterparts (Figure 1, lanes 4–9), with only monomeric, dimeric, and trimeric species present. After heating at 100°C for 20 min, followed by 60°C for 2 h in 20 mM Tris-HCl with 2% SDS at pH 4, 99% of monomeric protein was recovered as visualized by SDS–PAGE (Figure 3, lane 3).

The effect of NaBH3CN reduction on the formation of formaldehyde-induced cross-links. Lane M: molecular weight marker; lane 1: native RNase A reduced with NaBH3CN and incubated in 100% ethanol for 1 week; lane 2: formaldehyde-fixed RNase A after reduction with NaBH3CN and incubation in 100% ethanol for 1 week; lane 3: the sample from lane 2 after protein recovery in 20 mM Tris-HCl, pH 4.0, with 2% SDS. The RNase A samples were heated at 100°C for 20 min, followed by 60°C for 2 h.
Effects of Fixation and Ethanol Dehydration on Secondary and Tertiary Structure
We next examined the structural properties of native, formaldehyde-fixed, and formaldehyde-fixed, ethanol-treated RNase A using CD spectropolarimetry. RNase A is in the α+β structural class of proteins, with a secondary structure consisting of one long four-stranded antiparallel β-sheet and three short α-helixes.23 The solvent-corrected far-UV spectrum of native RNase A, which is sensitive to the secondary structure of the protein, is shown in Figure 4a, profile 1.10 The spectrum exhibits a minimum at ∼212 nm, with a broad shoulder centered at ∼220 nm, which is characteristic of an α+β protein conformation. Incubation of native RNase A in 10% formalin (Figure 4a, profile 3) for 1 week did not significantly alter the secondary structure of the protein. In addition, non-fixed RNase A incubated under ethanol for 1 week recovered its native structure after the ethanol was removed and the RNase A was reconstituted in phosphate buffer (Figure 4a, profile 2). However, when the formaldehyde-fixed RNase A was incubated under ethanol for 1 week and then rehydrated in phosphate buffer (Figure 4a, profile 4), there was a significant decrease in band intensity, and the profile changed to one with a single negative peak around 215 nm. This spectrum is characteristic of an all-β protein conformation.24 This spectrum was also observed for native, formaldehyde-fixed, and formaldehyde-fixed, ethanol-treated RNase A in 80% aqueous ethanol solution where all three proteins were minimally soluble (data not shown).

The effect of ethanol on protein structure: far-UV (a) and near-UV (b) CD spectra of 0.65 mg/ml solutions of RNase A. Profile 1: native RNAse A; profile 2: native RNase A incubated under 100% ethanol for 1 week and then rehydrated in phosphate buffer; profile 3: RNase A kept in 10% formaldehyde for 1 week; profile 4: RNase A fixed in 10% formaldehyde, incubated under 100% ethanol for 1 week, and then rehydrated in phosphate buffer.
The near-UV CD spectra of RNase A, which is sensitive to protein tertiary structure, are largely determined by six tyrosine residues,25,26 three of which (Tyr25, Tyr 92, and Tyr97) are buried within the protein interior. The remaining Tyr residues (Tyr73, Tyr76, and Tyr115) are located on the protein's surface. Non-formaldehyde-treated RNase A incubated in ethanol (Figure 4b, profile 2) or 10% formalin (Figure 4b, profile 3) for 1 week exhibited no significant changes in their near-UV CD spectra, as compared to native RNase A (Figure 4b, profile 1). However, there was a 60% decrease in negative band intensity for the formaldehyde-treated protein after prolonged exposure to ethanol (profile 4). This relative decrease may reflect the partial exposure of buried Tyr residues and/or an increase in the distance between Tyr73 and Tyr115 within the RNase A tertiary structure.25,27 These changes likely result from the collapse of tertiary structure as the spectrum is analogous to those seen in molten globule proteins28 and disordered proteins.24
DISCUSSION
Both native and formaldehyde-fixed RNase A undergo a structural transition from the native α+β to a nearly all-β conformation as ethanol concentration is increased to≥80%. The transition from the native to an all-β conformation at high ethanol concentrations is characteristic of most soluble proteins29,30,31 and is driven by the disruption of water structure by ethanol and the associated energetically unfavorable interaction of ethanol with the peptide backbone.32 The response of most proteins to this situation is to form β-sheets to sequester the peptide bonds away from the solvent while exposing nonpolar side chains to the alcohol.24 This secondary structural transformation is typically accompanied by a significant disruption (collapse) of tertiary structure33 as was observed for RNase A in the current study. This new protein conformation is further stabilized by the formation of intermolecular hydrogen bonds between geometrically compatible hydrophobic β-sheets,34 which then leads to extensive protein aggregation.30,35 Such β-sheet aggregates can form in response to a number of protein structural perturbants, in addition to alcohol, and are intermediates in the formation of amyloid fibrils by some proteins, including lysozyme, which, like RNase A exhibits an α+β structure in its native conformation.24,35,36 At ethanol concentrations≥80%, lysozyme formed amyloid-like protofibrils and exhibited a far-UV CD structure virtually identical to that observed for formaldehyde-fixed, ethanol-treated RNase A (Figure 4a), or native RNase in 80% aqueous ethanol (data not shown).24,35 Consequently, our results demonstrate that the presence of formaldehyde adducts neither inhibits nor facilitates the conformational changes exhibited by RNase A in the presence of ethanol. An important implication of the above observations is that formaldehyde-fixed RNase A maintains a high degree of conformational flexibility despite the presence of intra- and intermolecular formaldehyde cross-links. Thus, in the case of RNase A one can conclude that formaldehyde cross-links do not ‘lock-in’ either secondary or tertiary protein structure.
The protein aggregates formed by exposure of native RNase A to ethanol were reversible using the protein recovery conditions described in this study, whereas those formed by formaldehyde-fixed, ethanol-treated RNase A were only partially reversible. There are several possible explanations for this observation. One possible explanation is that the neutralization of charged amino acids by the formation of formaldehyde adducts contributes to aggregate formation by increasing the hydrophobicity of the protein surface. We have previously shown that formaldehyde treatment lowers the isoelectric point of RNase A from 9.2 to ∼7.4.10 Treatment of formaldehyde-fixed RNase A with NaBH3CN argues against this interpretation because this reagent reduces formaldehyde adducts to methyl groups, which further increases protein hydrophobicity. However, NaBH3CN treatment results in greater recovery of RNase A monomer (Figure 3), which is the opposite of what would be expected if the protein aggregates were stabilized predominantly by hydrophobic bonding.
Exposure of formaldehyde-fixed RNase A with PLP prior to ethanol treatment did not reduce the degree of protein oligomerization seen prior to protein recovery (Figure 2, lanes 2, 4, and 6). However, progressively more monomeric protein was recovered with increasing concentrations of PLP following high-temperature treatment (Figure 2, lanes 3, 5, and 7). This is interpreted to indicate that the reaction of PLP with the Schiff base or methylol adducts diminishes the number of formaldehyde cross-links formed, but does not inhibit the hydrogen bonding of the β-sheets. In contrast, reduction with NaBH3CN increased the fraction of monomeric protein seen by SDS–PAGE both before and after protein recovery by high-temperature treatment (Figure 3, lanes 2 and 3). Reduction of formaldehyde adducts prior to exposure to ethanol will clearly prevent cross-link formation, but likely also reduces hydrogen bonding through the reductive elimination of hydrogen-bond donors, such as lysine.7
Taken together, the above findings suggest that the cross-links in formaldehyde-treated RNase A in ethanol are difficult to reverse because they are largely sequestered within the intermolecular hydrophobic β-sheets present in the protein aggregates.
These cross-links may result from the rearrangement of existing cross-links or by the formation of new cross-links from latent formaldehyde adducts and formaldehyde-reactive amino-acid side chains present in the protein's interior. The latter may occur because the coplanar orientation of the side chains in β-sheets may provide a more favorable geometry for forming formaldehyde cross-links than the α-helix conformation (unpublished experiments). This conclusion is further supported by previous findings in which intermolecular cross-linking was increased by heating RNase A above its unfolding transition temperature in the presence of formaldehyde, indicating that additional cross-links were formed by previously buried formaldehyde adducts or formaldehyde-reactive amino-acid side chains.10 An additional contributing factor is that the dehydrating effects of ethanol may promote the conversion of methylol adducts to reactive Schiff-base intermediates that subsequently form cross-links that are less easily reversed than those formed by methylol adducts.7
In summary, the present study suggests that the ethanol-dehydration step in tissue histology is important in confounding the recovery of proteins from FFPE tissues. Ethanol-induced rearrangement of protein conformation can lead to protein aggregation through the formation of large geometrically compatible hydrophobic β-sheets that are stabilized by hydrogen bonds, formaldehyde cross-links, and van der Waals interactions. Such β-sheets would require substantial energy to induce sufficient hydration to reverse the formaldehyde cross-links within these sheets and regenerate protein monomers free of formaldehyde modifications. We have recently demonstrated that this required energy can be introduced using extremely high temperatures >100°C12 or by the application of elevated hydrostatic pressure at moderate temperatures.14
References
Chaurand P, Stoeckli M, Caprioli RM . Direct profiling of proteins in biological tissue sections by MALDI mass spectrometry. Anal Chem 1999;71:5263–5270.
Conti CJ, Larcher F, Chesner J, et al. Polyacrylamide gel electrophoresis and immunoblotting of proteins extracted from paraffin-embedded tissue sections. J Histochem Cytochem 1988;36:547–550.
de Noo ME, Deelder A, van der Werff M, et al. MALDI-TOF serum protein profiling for the detection of breast cancer. Onkologie 2006;29:501–506.
Cho WC, Cheng CH . Oncoproteomics: current trends and future perspectives. Expert Rev Proteomics 2007;4:401–410.
Neubauer H, Fehm T, Schutz C, et al. Proteomic expression profiling of breast cancer. Recent Results Cancer Res 2007;176:89–120.
Reymond MA, Schlegel W . Proteomics in cancer. Adv Clin Chem 2007;44:103–142.
Metz B, Kersten GFA, Hoogerhout P, et al. Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J Biol Chem 2004;279:6235–6243.
Metz B, Kersten GFA, Baart GJ, et al. Identification of formaldehyde-induced modifications in proteins: reactions with insulin. Bioconjug Chem 2006;17:815–822.
Crockett DK, Lin Z, Vaughn CP, et al. Identification of proteins from formalin-fixed paraffin-embedded cells by LC-MS/MS. Lab Invest 2005;85:1405–1415.
Rait VK, O’Leary TJ, Mason JT . Modeling formalin fixation and antigen retrieval with bovine pancreatic ribonuclease A: I—structural and functional alterations. Lab Invest 2004;84:292–299.
Rait VK, Xu L, O’Leary TJ, et al. Modeling formalin fixation and antigen retrieval with bovine pancreatic RNase A II. Interrelationship of cross-linking, immunoreactivity, and heat treatment. Lab Invest 2004;84:300–306.
Fowler CB, Cunningham RE, O’Leary TJ, et al. ‘Tissue surrogates’ as a model for archival formalin-fixed paraffin-embedded tissues. Lab Invest 2007;87:836–846.
Srinivasan M, Sedmak D, Jewell S . Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol 2002;161:1961–1971.
Fowler CB, Cunningham RE, Waybright TJ, et al. Elevated hydrostatic pressure promotes protein recovery from formalin-fixed, paraffin-embedded tissue surrogates. Lab Invest 2008;88:185–195.
Watkins NG, Thorpe SR, Baynes JW . Glycation of amino groups in protein. Studies on the specificity of modification of RNase by glucose. J Biol Chem 1985;260:10629–10636.
Maralihalli GB, Bhagwat AS . Interaction of pyridoxal phosphate with the amino groups at the active site of 5-aminolevulinic acid dehydratase in maize. J Biosci 1985;7:359–364.
Shi S-R, Liu C, Balgley BM, et al. Protein extraction from formalin-fixed, paraffin-embedded tissue sections: quality evaluation by mass spectrometry. J Histochem Cytochem 2006;54:739–743.
Brewer J, Pesce A, Ashworth R . Experimental Techniques in Biochemistry. Prentice Hall Inc: Englewood Cliffs, NJ, 1974.
Bratthauer GL . Processing of tissue specimens. Methods Mol Biol 1999;115:77–84.
Hall JA, Maloney PC . Pyridoxal 5-phosphate inhibition of substrate selectivity mutans of UhpT, the sugar 6-phosphate carrier of Escherichia coli. J Bacteriology 2002;184:3756–3758.
Mason JR, Leong FC, Plaxco KW, et al. Two-step covalent modification of proteins. Selective labeling of Schiff base-forming sites and selective blockade of the sense of smell in vivo. J Am Chem Soc 1985;107:6075–6084.
Borch RF, Bernstein MD, Durst HD . Cyanohydridoborate anion as a selective reducing agent. J Am Chem Soc 1971;93:2897–2904.
Wlodawer A, Bott R, Sjölin L . The refined crystal structure of ribonuclease A at 2.0 A resolution. J Biol Chem 1982;257:1325–1332.
Cao A, Hu D, Lai L . Formation of amyloid fibrils from fully reduced hen egg white lysozyme. Protein Sci 2004;13:319–324.
Woody RW, Dunker AK . Aromatic and cysteine sidechain circular dichroism in proteins. In: Fasman GD (ed). Circular Dichroism and the Conformational Analysis of Biomolecules. Plenum Press: New York, 1996,pp 109–157.
Stelea SD, Pancoska P, Benight AS, et al. Thermal unfolding of ribonuclease A in phosphate at neutral pH: deviations from the two-state model. Protein Sci 2001;10:970–978.
Katakura Y, Kumamoto T, Iwai Y, et al. Fluorescence polarization study of a salt bridge between a single-chain Fv and its antigen ribonuclease A. Mol Immunol 1997;34:731–734.
Peng Z, Kim PS . A protein dissection study of a molten globule. Biochemistry 1994;33:2136–2141.
Dobson CM . Protein misfolding, evolution, and disease. Trends Biochem 1999;24:329–332.
Tanaka S, Oda Y, Ataka M, et al. Denaturation and aggregation of hen egg lysozyme in aqueous ethanol solution studies by dynamic light scattering. Biopolymers 2001;59:370–379.
Yan YB, Zhang J, He HW, et al. Oligomerization and aggregation of bovine pancreatic ribonuclease A: characteristic events observed by FTIR spectroscopy. Biophys J 2006;90:2525–2533.
Pace CN, Trevino S, Prabhakaran E, et al. Protein structure, stability and solubility in water and other solvents. Philos Trans R Soc Lond B Biol Sci 2004;359:1225–1235.
Kamatari YO, Konno T, Kataoka M, et al. The methanol-induced transition and the expanded helical conformation in hen lysozyme. Protein Sci 1998;7:681–688.
Herskovits TT, Jaullet H . On the structural stability and solvent denaturation of proteins. J Biol Chem 1970;245:2588–2598.
Goda S, Takano K, Yamagata Y, et al. Amyloid protofilament formation of hen egg lysozyme in highly concentrated ethanol solution. Protein Sci 2000;9:369–375.
Guijarro JI, Sunde M, Jones JA, et al. Amyloid fibril formation by an SH3 domain. Proc Natl Acad Sci USA 1998;95:4224–4228.
Acknowledgements
This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Grant 1R33 CA 107844. The content of this publication does not necessarily reflect the views or policies of the Department of Defense, or the Veterans Health Administration, nor does mention of trade names, commercial products, or organization imply endorsement by the United States Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fowler, C., O'Leary, T. & Mason, J. Modeling formalin fixation and histological processing with ribonuclease A: effects of ethanol dehydration on reversal of formaldehyde cross-links. Lab Invest 88, 785–791 (2008). https://doi.org/10.1038/labinvest.2008.43
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.43
