
Abstract
Four new angucyclin(on)es, 11-deoxylandomycinone (1) and landomycins X–Z (2–4) were isolated from the crude extract of Streptomyces cyanogenus K62 mutant strain, along with the recently reported landomycins S, T and V (5–7) and five other known compounds. The structures of the new compounds 1–4 were elucidated by 1D and 2D NMR studies along with HR-MS analyses. Unique about the structures is that the fourth sugar moiety (sugar D) in landomycins X–Z (2–4) was β-D-amicetose instead of β-D-olivose, usually found in this position. The new angucyclin(on)es were biologically evaluated in comparison with previously known congeners against a small panel of MCF-7 (estrogen responsive) and MDA 231 (estrogen refractory) breast cancer cell lines. 11-deoxylandomycinone (IC50 2.1±0.3 and 1.2±0.4 μM) and landomycin Y (IC50 1.0±0.1 and 2.0±0.1 μM) showed the highest cytotoxic potencies against both the cell lines.
Similar content being viewed by others

Introduction
The landomycins are a subgroup of the large family of angucycline group antibiotics, which are characterized by diverse biological activities, such as antitumor, antibacterial and enzyme inhibition.1, 2, 3, 4, 5, 6, 7 The chemical structures of the landomycins consist of a polyketide-derived angucyclinone decorated with a single deoxyoligosaccharide chain of various lengths. Landomycins A–D (C, 13) were originally found as products of Streptomyces cyanogenus S136.3, 5, 6 Later, several more landomycins were discovered, and analyzed for their structure–activity relationships.8, 9, 10, 11, 12 It was found that the biological activities were mainly dependent on the length of the saccharide chain, with those analogs possessing longer saccharide chains, being more potent in general.10, 13, 14 Landomycin A, the principal product of S. cyanogenus S136, is the most potent antitumor agent, possessing an unusual spectrum of activity against the NCI 60 human cancer cell line panel.1, 2, 6 It contains a hexasaccharide side chain, constructed from two repeating trisaccharide patterns (D-olivose-4-1-D-olivose-3-1-L-rhodinose). Except for landomycin C (13), the sugar chains of all reported landomycins are constructed solely from L-rhodinose and D-olivose units. Landomycin C (13) was the only analog that bears three different sugar moieties; D-olivose, L-rhodinose and D-amicetose.
During our search for further new cytotoxic landomycin analogs, a fermentation of S. cyanogenus K62 in SG-medium was carried out which afforded four new angucyclin(on)es: 11-deoxylandomycinone (1) and landomycins X–Z (2–4) along with the known compounds tetrangulol (11), tetrangomycin (12), landomycins M (8), F (9) and O (10).10, 11, 15, 16, 17 In addition, we also found again the very recently reported landomycins S, T and V (5–7) (Scheme 1).12
Results and discussion
In our search for new landomycin analogs with altered saccharide patterns we screened the regulator-affected high producing mutants S. cyanogenus K62 and S. cyanogenus K60.18 The production spectrum using SG-medium was very similar for both mutant strains, based on the TLC and HPLC-MS analyses (Supplementary Figure S1). However, the general production yields of S. cyanogenus K62 were significantly higher than those of S. cyanogenus K60. Therefore, we focused on the K62 mutant for the search of new minor congeners.
A pre-culture of S. cyanogenus K62 served to cultivate 40 of 0.25 L-Erlenmeyer flasks each containing 100 ml of SG-medium, on rotary shaker for 3 days. The broth was harvested, mixed with celite, filtered off and extracted with ethyl acetate, and the organic extracts from supernatant and cells were concentrated in vacuo to afford 6.45 g of a reddish powder crude extract (1.61 g l−1). A TLC analysis of the strain extract exhibited several UV orange-red fluorescent bands at 366 nm, which turned blue on treatment with 2 N NaOH, as indicative of peri-hydroxy quinones. The HPLC-MS analysis of the crude extract displayed several components with UV spectrum characteristic of 11-deoxylandomycin chromophores, which were likely new congeners (Supplementary Figures S4 and S5).12 Working up and purification of 0.97 g from the strain extract using various chromatographic techniques (Figure 1) led to the isolation of four new compounds; 11-deoxylandomycinone (1) and landomycins X-Z (2–4), all three possessing 11-deoxyaglycone moiety. In addition, eight known compounds; tetrangulol (11), tetrangomycin (12), landomycins M, F and O (8–10) were isolated along with the recently reported landomycins S, T and V (5–7).

Work-up procedure of extracts from Streptomyces cyanogenus K62.
Structure elucidation
Compound 1 was obtained as an orange amorphous powder. The molecular formula of 1 was determined by high resolution electrospray ionization mass spectrometry (HRESIMS) as C19H14O5 (Tables 1 and 2). The proton NMR spectrum of 1 displayed two broad singlets at δ 12.07 and 9.75, representing peri-hydroxy groups, and a 1,2,3-trisubstituted aromatic moiety revealed by an ABC system in the region of δ 7.73∼7.32 (J=7.5∼8.8 Hz, Table 3). Two additional broad aromatic signals, each 1H, at δ 6.64 and 6.57 showed another highly substituted aromatic ring with two m-coupled aromatic protons. The aliphatic region revealed an oxymethine signal (δ 5.03) directly next to a methylene group (δ 2.89 and 2.76; d, J=16.2∼16.2 Hz), which was confirmed by a H,H-COSY experiment (Figure 2). Furthermore, a singlet of an aromatic-bound methyl group was observed at δ 2.26. All these structural features are typical for 11-deoxylandomycinone. The 13C NMR/hetero single quantum coherence (HSQC) spectra (Table 4) confirmed compound 1 to be 11-deoxylandomycinone, and showed the quinone carbonyls (δ 188.0 and 183.7), the small Δδ∼4 p.p.m. indicating both carbonyls to be chelated with hydroxyl groups. In the sp3 region, the three expected carbon signals representing an oxymethine carbon (δ 57.1), methylene (δ 36.5) and one methyl (δ 21.2) groups, were observed. Finally, the HMBC spectrum (Figure 2) of compound 1 showing 3J correlations between H-11 and C-12, and between H-6 and C-7, confirmed structure 1 as 11-deoxylandomycinone, with C-6 being R-configured, as it displayed the same coupling constants and NOESY correlations (Figure 2 and Table 3) typically of all reported landomycins. A data base search (Chemical Abstracts) confirmed the novelty of structure 1.

HMBC connectivities ( → ), H,H COSY correlations (bold lines) and diagnostic NOESY couplings (↔) of 11-deoxylandomycinone (1).
Compound 2 was obtained as orange solid, with a molecular weight of 1054 Da corresponding to a molecular formula of C55H74O20, as deduced by HRESIMS (Tables 1 and 2). The proton NMR spectrum (Table 3) and the 13C NMR/HSQC spectra (Table 4) of 2 showed that it contains an 11-deoxylandomycinone agylcone, plus six saccharide moieties (six anomeric 1H, δH 5.18−4.41; δC 103.7−97.5). Four of the anomeric protons (δ 5.18 dd, J=9.5, 1.5 Hz; δ 4.51 dd, J=8.6, 1.3 Hz; δ 4.48 dd, J=9.8, 1.3 Hz; δ 4.41 dd, J=7.9, 1.3 Hz) show large coupling constants and thus represent β-D-glycoside moieties. The remaining two anomeric protons at δ 4.94 (brs) and δ 4.92 (brs) are α-glycosidically linked L-sugars. The 2D-NMR studies revealed that all these sugars are part of one hexasaccharide chain, linked—as with all landomycins—at 8-position. Overall, structure 2 most closely resembled the recently discovered landomycin S (5), however, it is smaller by 16 amu, because of the lack of one oxygen atom in the hexasaccharide side chain, as revealed by the comparison of MS/MS fragmentation patterns of 2 with those of landomycin S (5) (Supplementary Figure S3). The NMR (H,H-COSY and HMBC correlations) and MS data analysis showed that the difference was in the fourth sugar moiety, with sugar D being a D-amicetose instead of the D-olivose unit usually found in this position. The data also proved the attachment of the hexasaccharide at 8-position (3JC−H coupling between H-1A, δH 5.18 with C-8, δC 156.4), for MS/MS fragmentation see also Supplementary Figure S3. All of the remaining NMR data (Tables 3 and 4 and Figure 3) are in full agreement with structure 2. The relative configurations of the sugar residues were derived from the coupling constants and NOESY experiments (Figure 4) indicating that compound 2 has the same stereochemistry both at C-6 of the aglycone and the hexasaccharide sugar moieties as found previously for landomycin C (13). In sum, structure 2 was determined to be 11-deoxylandomycin C, and was named landomycin X.

Selected HMBC connectivities ( → ) and H,H COSY correlations (bold lines) of Landomycin X (2).

Diagnostic NOESY correlations (↔) of Landomycin X (2).
Closely related to landomycin X (2), compound 3 was obtained as dark red solid from the same fraction III, exhibiting a molecular formula of C55H72O19 (HRESI MS), which is by 18 amu (one H2O) smaller than the one of landomycin X (2), and has one more degree of unsaturation (Tables 1 and 2). The 1H and 13C NMR data of 3 were similar to those of 2 (Tables 3 and 4), except that ring B of the aglycone was aromatic, as revealed by the 1H NMR spectrum (two additional ortho-coupled protons at δ 8.08 (d, J=8.8 Hz) and 8.23 (d, J=8.6) of 3. The structure of 3 was additionally deduced by H,H-COSY, HSQC, HMBC and NOESY experiments, exhibiting the same structural and stereochemical features as in compound 2 (Figures 5 and 6). Therefore, structure 3 was determined as 5,6-anhydro-landomycin X, and consequently named landomycin Y.

Selected HMBC connectivities ( → ) and H,H COSY correlations (bold lines) of Landomycin Y (3).

Diagnostic NOESY correlations (↔) of Landomycin Y (3).
Structurally related to landomycin X (2) and the recently reported landomycin V (7)12, compound 4 was obtained as orange solid, with a molecular formula of C49H64O18 (HREIMS); that is, by 16 amu smaller than landomycin V (7), for physico-chemical properties see Tables 1 and 2. Comparison of the 1H NMR data of compound 4 with those of landomycin X (2) revealed that the terminal α-L-rhodinose moiety was missing, whereas the aglycone was identical to the one found in compounds 2 and 7. Compared with structure 7 an oxygen atom was missing in compound 4, again at position 3D, due a D-amicetose unit instead of a D-olivose (H,H-COSY correlations, Supplementary Figure S2, Table 3). Thus, compound 4 was identified as 3D-deoxy-landomycin V, and consequently named landomycin Z.
Biological activity
The anticancer activity of the new angucyclin(on)es 1–4 compared with landomycin A were determined using MCF-7 (estrogen responsive) and MDA 231 (estrogen refractory) breast cancer cells (Table 5). Cell viability assays showed that compounds 1–4 and landomycin A have comparable anticancer activities against both cells lines. Specifically, against MCF-7 cells, compound 3 was the most potent (IC50=1.0 μM), but also compounds 1, 2 and 4 appear to have comparable activity (IC50=2.1, 2.8 and 2.6 μM, respectively) to landomycin A. 11-deoxylandomycinone (1) (IC50=1.2 μM) was the most potent compound against MDA 231 cells. However compounds 2–4, (IC50=2.0, 2.0 and 2.5 μM, respectively) also displayed significant cytotoxic activities, again comparable to landomycin A. In conclusion, unlike some of the previously discovered new 11-deoxy-landomycins; for example, landomycins F (9), M (8), S (5), T (6) and V (7), the new angucylin(on)es 1–4 showed potency against both MDA 231 and MCF-7 cells, previously only found for landomycin A and other landomycins bearing an 11-OH group. The exchange of the fourth sugar moiety (β-D-olivose) of landomycins S, T and V (5–7) with β-D-amicetose as in the new landomycins X–Z (2–4) slightly improve the anticancer activity (Table 5). The results suggest that a missing 4D-OH group; that is, substitution of D-olivose by a D-amicetose unit in D-position of the saccharide chain, is advantageous, showing that subtle changes in the H-bonding properties of the saccharide chains can have a significant effect. As discussed before, the highest activity of landomycins X–Z (2–4) and aglycone 1 indicate that these compounds may have different mechanism-of-action, one for the aglycone alone, the other depending on the length of the sugar side chain, again with longer chains being advantageous. It should also be noted that the observed effects on estrogen receptor (ER)-negative (MDA-231) compared with ER-positive (MCF-7) breast cancer cells could be influenced by differential gene expression patterns known from these cell lines; for example, MDA-231 cells express higher cdc2, cyclin B1, cyclin D1, cyclin E, IGFBP-3, TGF-α, TGFβ2 compared with MCF-7 cells. Investigations of the molecular mechanism of the landomycins are currently in progress.
Experimental procedure
General experimental procedures
UV spectra were recorded on a Shimadzu UV-1800 (Model TCC-240A) UV spectrometer. NMR spectra were measured on a Varian VnmrJ 500 (1H, 500 MHz; 13C, 125.7 MHz) spectrometer, the δ-values were referenced to the respective solvent signals. ESI mass spectra were recorded on a Finnigan LCQ ion trap mass spectrometer. Electrospray ionization high resolution mass spectra were recorded on an Agilent LC/MSD TOF (resolution: 10 000; 3 p.p.m. mass accuracy; Inlet Systems: Agilent Technologies 1200 Series LC pumps) Mass Spectrometer (Agilent, Palo Alto, CA, USA). LC/MS/MS measurements were performed on an Applied Biosystems 3200 QTRAP mass spectrometer (Applied Biosystems, Foster City, CA, USA) using electrospray ionization in the positive and negative ionization mode, inlet systems: Agilent 1100 series HPLC; Resolution: Unit mass. Samples were introduced by means of a syringe pump. HPLC purifications were carried out using a Symmetry Prep C1810 μm column (10 × 150 mm) on a binary LC system. HPLC-MS analyses were carried out using a Symmetry Anal C185 μm column (4.6 × 250 mm) on a binary LC system. Flash chromatography was carried out on silica gel MN 60 (140–270 mesh, American Society of Testing Materials). Rf values were measured on Polygram SIL G/UV254 (Macherey-Nagel, Dueren, Germany). Size-exclusion chromatography was performed on Sephadex LH-20 (GE Healthcare, Piscataway, NJ, USA).
Cell viability assay
To determine the cytotoxic activity of the new compounds 11-deoxylandomycinone (1), landomycins X–Z (2–4) and landomycin A were tested against two breast cancer cell lines, MCF-7 (estrogen responsive) and MDA 231 (estrogen refractory). Cell viability of these two cell lines in response to the various concentrations of compounds were determined using the trypan blue exclusion assay, in which 50 × 103 cells in 0.5 ml medium were plated in each well of a 24-well plate and allowed to attach overnight. The medium was replaced the following day with fresh medium containing different concentrations of the compounds to be tested and the plates were incubated for 24 h at 37 °C. At the end of the treatment period both adherent and floating cells were collected, and resuspended in phosphate-buffered saline for trypan blue staining using 0.4% stain for 3 min. Stained (dead) and unstained (live) cells were counted using a hemocytometer, cell viability in response to specific compounds was determined, dose–response curve was plotted and finally IC50 was calculated. Each set of experiment was performed three times to confirm reproducibility of the results.
Culture material, fermentation and isolation
SG-Medium
Glucose (20 g, Sigma-Aldrich, St Louis, MO, USA), yeast extract (5 g, Acros Organics, Morris Plains, NJ, USA), Soytone (10, Becton, Dickinson, Franklin Lakes, NJ, USA), CoCl2 x 6 H2O (1 mg, Acros Organics, Morris Plains, NJ, USA) and calcium carbonate (2 g, Sigma-Aldrich) were dissolved in 1 l of demineralized water. The suspension (pH 7.2) was sterilized by autoclaving for 33 min at 121 °C.
M2-Agar Medium
Glucose (4.0 g, Sigma-Aldrich), yeast extract (4.0 g, Acros Organics), malt extract (10.0 g, MP Biomedicals, LLC, Solon, OH, USA) and agar (15.0 g, Becton, Dickinson) were dissolved in 1 l of demineralized water.
Fermentation, extraction and isolation
Strain S. cyanogenus K62 was cultivated on M2-agar plates at 28 °C for 2 days. With pieces of well-grown agar subculture of the strain, a pre-culture (0.25 L-Erlenmeyer flask) of S. cyanogenus K62, containing 100 ml of SG-medium was prepared, inoculated and cultivated at 28 °C (250 r.p.m.). After 2 days the grown pre-culture flask was used to inoculate 40 of the 0.25 l flasks each containing 100 ml of SG-medium, which was grown at 28 °C, and harvested after 3 days. The obtained reddish brown culture broth was mixed with celite and filtered off; both biomass and filtrate were extracted with EtOAc; (5 × 500 ml, for biomass) and (4 × 2 l, for filtrate). Both extracts were combined and evaporated in vacuo at 40 °C, and afforded 6.45 g of reddish powder crude extract.
Separation of 0.97 g of crude extract on silica gel column (column 2.5 × 50 cm, 100 g), using a stepwise MeOH/CH2Cl2 gradient (0.2 l 0% MeOH → fraction FI, then 0.2 l 5% MeOH → fraction FII, then 0.2 l 10% MeOH, then 0.5 l 50% MeOH, combined → fraction FIII), yielded three fractions, FI (100 mg, red solid), FII (60.7 mg, orange solid) and FIII (570 mg, red solid). Fraction FI was further purified during silica gel column (0.5 l, CH2Cl2/20% n-hexane; 2 × 30 cm) followed by Sephadex LH-20 (2 × 40 cm, 50% MeOH/CH2Cl2) to obtain tertangulol (11; reddish brown crystals, 38.2 mg). Purification of fraction FII was carried out by HPLC followed by Sephadex LH-20 (1 × 20 cm, MeOH) to yield tetrangomycin (12; yellow solid, 1.3 mg) and 11-deoxylandomycinone (1; orange solid, 6.1 mg,). In an analogous manner, further fractionation and purification of fraction FIII delivered landomycins F (9, 60.0 mg), O (10, 37.1 mg), V (7, 24.9 mg), S (5, 38.7 mg), M (8, 15.8 mg), T (6, 31.2 mg), along with the three new landomycins X∼Z (2–4, 11.6, 9.39 and 2.1 mg, respectively) in pure form, (Figure 1, Supplementary Figure S4).
11-Deoxylandomycinone (1)
Orange solid; Rf 0.87 (7% MeOH/CH2Cl2), blue coloration with 2N NaOH; UV (MeOH) λmax (log ɛ) 263 (3.71), 288 (3.68), 319 sh (3.58), 447 (3.28) nm; 1H NMR (DMSO-d6, 500 MHz) δ 12.07 (1H, brs, 8-OH), 9.75 (1H, brs, 1-OH), 7.73 (1H, t, J=8.8 Hz, H-10), 7.44 (1H, d, J=7.5 Hz, H-11), 7.32 (1H, d, J=8.8 Hz, H-9), 6.64 (1H, brs, H-2), 6.57 (1H, brs, H-4), 5.03 (1H, d, J=3.9 Hz, 6-OH), 4.97 (1H, brs, H-6), 2.89 (1H, d, J=16.2 Hz, Hβ-5), 2.76 (1H, d, J=16.4 Hz, Hα-5), 2.26 (3H, s, 3-CH3) p.p.m.; 1H NMR (CDCl3, 500 MHz) and 13C NMR (DMSO-d6, 125 MHz), see Tables 3 and 4; (−)-ESI MS m/z 321 [M-H]–; (+)-ESI MS m/z 323 [M+H]+; (−)-HRESIMS m/z 321.0768 [M-H]– (calcd for C19H13O5, 321.0768); (+)-HRESIMS m/z 323.1001 [M+H]+, 305.0795 [M-H2O+H]+, 361.0473 [M+K]+ (calcd for C19H15O5, 323.0914, for C19H13O4, 305.0808 and for C19H14O5K, 361.0473).
Landomycin X (2)
Orange solid; Rf 0.65 (7% MeOH/CH2Cl2), blue coloration with 2N NaOH; UV (MeOH) λmax (log ɛ) 265 (4.41), 285 (4.35), 320 sh (4.13), 412 (3.93) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 3 and 4; (−)-ESI MS m/z 1053 [M-H]–; (+)-ESI MS m/z 1077 [M+Na]+; (–)-ESI MS/MS m/z (%) 1053 ([M-H]−, 100), 1035 ([M-H2O-H]−, 5), 893 (70), 321 ([M-(-(L-rhodinose + D-olivose + D-amicetose + L-rhodinose + D-olivose + D-olivose)-H]−, 50); (−)-HRESIMS m/z 1053.4688 [M-H]− (calcd for C55H73O20, 1053.4700); (+)-HRESIMS m/z 1077.4722 [M+Na]+, 1093.4467 [M+K]+ (calcd for C55H74O20 Na, 1077.4665 and for C55H74O20K, 1093.4405).
Landomycin Y (3)
Dark red solid; Rf 0.60 (7% MeOH/CH2Cl2), blue coloration with 2N NaOH; UV (MeOH) λmax (log ɛ) 246 sh (4.59), 312 (4.59), 399 (4.03) nm; 1H NMR (CDCl3,500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 3 and 4; (+)-ESI MS m/z 1059 [M+Na]+; (+)-HRESIMS m/z 1059.4546 [M+Na]+ (calcd for C55H72O19Na, 1059.4560).
Landomycin Z (4)
Orange solid; Rf 0.35 (7% MeOH/CH2Cl2), blue coloration with 2N NaOH; UV (MeOH) λmax (log ɛ) 265 (4.14), 285 (4.05), 403 (3.79) nm; 1H NMR (CDCl3, 500 MHz), see Table 3; (+)-ESI MS m/z 963 [M+Na]+; (+)-HRESIMS m/z 963.3981 [M+Na]+ (calcd for C49H64O18Na, 963.3985).

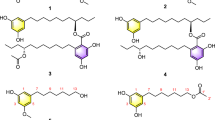
Chemical structures of the new landomycins and of related angucyclin(on)es.
References
Crow, R. T. et al. Landomycin A inhibits DNA synthesis and G1/S cell cycle progression. Bioorg. Med. Chem. Lett. 9, 1663–1666 (1999).
Depenbrock, H. et al. Assessment of antitumor activity of landomycin A (NSC 6399187-A). Ann. Hematol. 73 (Supl II) A80/316 (1996).
Rohr, J. & Thiericke, R. Angucycline group antibiotics. Nat. Prod. Rep. 9, 103–137 (1992).
Krohn, K. & Rohr, J. Angucyclines: total syntheses, new structures, and biosynthetic studies of an emerging new class of antibiotics. Topics Curr. Chem. 188, 127–195 (1997).
Weber, S., Zolke, C., Rohr, J. & Beale, J. M. Investigations of the biosynthesis and structural revision of landomycin A. J. Org. Chem. 59, 4211–4214 (1994).
Henkel, T., Rohr, J., Beale, J. M. & Schwenen, L. Landomycins, new angucycline antibiotics from Streptomyces sp. I. Structural studies on landomycins A-D. J. Antibiot. 43, 492–503 (1990).
Korynevska, A. et al. Mechanisms underlying the anticancer activities of the angucycline landomycin E. Biochem. Pharmacol. 74, 1713–1726 (2007).
Zhu, L. et al. Generation of new landomycins with altered saccharide patterns through over-expression of the glycosyltransferase gene lanGT3 in the biosynthetic gene cluster of landomycin A in Streptomyces cyanogenus S-136. Chem. Bio.Chem. 8, 83–88 (2007).
Zhu, L. et al. Identification of the function of gene lndM2 encoding a bifunctional oxygenase-reductase involved in the biosynthesis of the antitumor antibiotic landomycin E by Streptomyces globisporus 1912 supports the originally assigned structure for landomycinone. J. Org. Chem. 70, 631–638 (2005).
Luzhetskyy, A. et al. Generation of novel landomycins M and O through targeted gene disruption. Chem. Bio. Chem. 6, 675–678 (2005).
Ostash, B. et al. Generation of new landomycins by combinatorial biosynthetic manipulation of the LndGT4 Gene of the landomycin E cluster in S. globisporus. Chem. Biol. 11, 547–555 (2004).
Shaaban, K. A., Srinivasan, S., Kumar, R., Damodaran, C., Rohr, J. & Landomycins, P.- W. Cytotoxic angucyclines from streptomyces cyanogenus S13. J. Nat. Prod. submitted (2010).
Luzhetskyy, A., Vente, A. & Bechthold, A. Glycosyltransferases involved in the biosynthesis of biologically active natural products that contain oligosaccharides. Mol. BioSyst. 1, 117–126 (2005).
Trefzer, A. et al. Elucidation of the function of two glycosyltransferase genes (lanGT1 and lanGT4) involved in landomycin biosynthesis and generation of new oligosaccharide antibiotics. Chem. Biol. 8, 1239–1252 (2001).
Kuntsmann, M. P. & Mitscher, L. A. The structural characterization of tetrangomycin and tetrangulol. J. Org. Chem. 31, 2920–2925 (1966).
Krohn, K., Boker, N., Florke, U. & Freund, C. Synthesis of Angucyclines. 8. Biomimetic-Type synthesis of rabelomycin, tetrangomycin, and related ring B aromatic angucyclinones. J. Org. Chem. 62, 2350–2356 (1997).
Krohn, K. & Khanbabaee, K. First total synthesis of (+/−)-rabelomycin. Angew. Chem. Int. Ed. Engl. 33, 99–100 (1994).
Ostash, I. et al. Coordination of export and glycosylation of landomycins in Streptomyces cyanogenus S136. FEMS Microbiol. Lett. 285, 195–202 (2008).
Acknowledgements
The mass spectrometry department, University of Wisconsin Biotechnology Centre is acknowledged for the HR-MS data. This work was supported by grant CA 102102 from the US National Institutes of Health to JR.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Shaaban, K., Stamatkin, C., Damodaran, C. et al. 11-Deoxylandomycinone and landomycins X-Z, new cytotoxic angucyclin(on)es from a Streptomyces cyanogenus K62 mutant strain. J Antibiot 64, 141–150 (2011). https://doi.org/10.1038/ja.2010.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.121
Keywords
This article is cited by
-
Landomycin biosynthesis and its regulation in Streptomyces
Applied Microbiology and Biotechnology (2019)
-
Structure and biosynthesis of mayamycin B, a new polyketide with antibacterial activity from Streptomyces sp. 120454
The Journal of Antibiotics (2018)
-
Generation of new compounds through unbalanced transcription of landomycin A cluster
Applied Microbiology and Biotechnology (2016)
-
Langkocyclines: novel angucycline antibiotics from Streptomyces sp. Acta 3034
The Journal of Antibiotics (2013)
-
Warkmycin, a novel angucycline antibiotic produced by Streptomyces sp. Acta 2930
The Journal of Antibiotics (2013)
