
Abstract
The kidney has a central role in long-term control of blood pressure, and decreased kidney function is a common but difficult-to-treat cause of hypertension. Conversely, elevated blood pressure contributes to the progression of chronic kidney disease. Steroid hormone aldosterone and its receptor mineralocorticoid receptor (MR) contribute to hypertension by increasing renal salt reabsorption and promote kidney dysfunction through direct effects on renal parenchymal cells. Accumulating data indicate that various mechanisms affect aldosterone-MR signaling. Using a genetically engineered mouse model, we identified crosstalk between small GTPase Rac1 and MR. This crosstalk pathway promotes glomerular podocyte injury, and is also involved in the pathogenesis of hypertension. Notably, salt loading increases renal Rac1 activity in several models of salt-sensitive hypertension, which, in the presence of aldosterone, synergistically activates MR signaling, causing hypertension and kidney injury. There is also a mechanism regulating MR in a cell-selective manner. In the principal cells of the collecting duct, aldosterone directly binds and activate MR. In neighboring intercalated cells, however, binding of aldosterone to MR is regulated by phosphorylation at the ligand-binding domain. This mechanism serves as a switch to turn on electrolyte flux pathways in intercalated cells, allowing aldosterone to exert distinct effects in different physiological contexts. Given the potential benefit of MR blockade in hypertensive kidney disease, the delineation of these pathways may lead to the identification of alternative therapeutic targets. In this review, we discuss the roles of MR in mediating kidney disease and hypertension, with a focus on the crosstalk among related signaling pathways.
Similar content being viewed by others


Main
The kidney fluid system is a predominant regulator of blood pressure, and reduced kidney function constitutes a major cause of hypertension.1, 2, 3 High blood pressure, in turn, contributes to the progression of kidney dysfunction,4, 5 causing a vicious cycle in hypertensive kidney disease. Mineralocorticoid receptor (MR; encoded by NR3C2) belongs to the nuclear receptor superfamily and is abundantly present in the so-called aldosterone-sensitive distal nephron.6, 7 In response to the steroid hormone aldosterone, MR regulates the transcription of target genes, serving as the master regulator of Na+, K+ and Cl− flux mechanisms in this segment.8, 9 Among the various genes that cause Mendelian forms of blood pressure variation in humans, mutations in aldosterone synthase, MR and epithelial Na+ channel (ENaC) can cause both hypertension and hypotension,3 demonstrating the central role of the aldosterone-MR system in fluid volume homeostasis.
Besides its role in regulating blood pressure, MR has also been shown to be involved in the pathogenesis of end-organ damage, including heart failure and chronic kidney disease (CKD) progression.10, 11, 12, 13, 14, 15, 16 Especially, experimental and clinical studies have demonstrated that MR blockade effectively reduces proteinuria.14, 16, 17, 18, 19 It is now accepted that MR is much more widely distributed than previously thought,20, 21, 22, 23, 24 and aberrant MR signaling in nonclassical targets is likely to be involved in target organ damage associated with hypertension. Given the wide distribution of MR in various tissues and organs,20, 21, 22, 23, 24 it is plausible that the function of MR is locally modulated in a context-dependent manner. Accordingly, we identified previously unrecognized mechanisms that regulate MR function independently of circulating ligand levels.18, 22, 25 In this short review, we discuss the roles of MR in mediating kidney disease and hypertension, with a focus on the crosstalk among various related signaling pathways.
Potentiation of MR signaling by the small GTPase Rac1 and glomerular epithelial cell damage
The Rho family of small G proteins, which includes RhoA, Rac1 and Cdc42, controls cell shape, polarity and migration through actin reorganization.26 When activated, Rho family GTPases dissociate from the negative regulators, Rho GDP dissociation inhibitors (RhoGDIs), translocate to the cell membrane, and are converted to the active, GTP-bound form though interaction with guanine nucleotide-exchange factors. They are then converted to the inactive GDP-bound form by GTPase-activating proteins and bound to RhoGDIs. Interestingly, mice lacking RhoGDI-α (Arhgdia−/−) exhibit heavy proteinuria and progressive kidney failure,27 highlighting the critical role of this protein in kidney function. Using this mice model, we analyzed the mechanisms of the kidney injury in detail, and unexpectedly found that the activity of Rac1 but not that of RhoA was elevated,18 and pharmacological manipulation of Rac1 activity in the kidney revealed its pathological importance in glomerular podocyte injury and proteinuria.
In addition to its well-described role in actin cytoskeletal reorganization, Rac1 is also capable of regulating the nuclear accumulation and transcriptional activity of nuclear transcription factors.28, 29 In an effort to clarify the signaling pathways that cause kidney dysfunction in the Arhgdia−/− model, we found that Rac1 facilitates MR signaling independently of the ligand aldosterone levels.18 Indeed, Arhgdia−/− mice showed MR accumulation in the nuclear fraction and increased expression of MR target genes in the absence of hyperaldosteronism. Furthermore, these changes, in addition to heavy proteinuria and kidney dysfunction, were almost completely reversed by the selective MR antagonist eplerenone, demonstrating the central role of aberrant MR signaling. Thus, in this model, Rac1 activity in the kidney is increased by deletion of RhoGDIα, which in turn induces MR signaling by facilitating receptor function, thereby causing kidney dysfunction.
Interestingly, recent studies have identified and characterized a type of congenital nephrotic syndrome caused by mutations in RhoGDIα in humans.30, 31 Consistent with our data, the study showed that mutations in ARHGDIA (R120X, G173V) result in the activation of Rac1 but not that of RhoA. In addition, the nephrotic phenotype recapitulated in RhoGDIα-deficient zebrafish is ameliorated by Rac1 inhibitors and MR antagonists.30
Role of the Rac1-MR pathway in the pathophysiology of CKD associated with hypertension
The pressor response to high salt intake (termed ‘salt sensitivity’) varies among individuals;32 however, the related mechanisms have not been completely elucidated. Dahl salt-sensitive (Dahl-S) and salt-resistant (Dahl-R) strains have long been used to evaluate the factors influencing salt sensitivity. In these models, previous studies have reported that plasma aldosterone levels are rather low in Dahl-S rats compared with those in Dahl-R rats.33 Moreover, salt loading decreases aldosterone secretion similarly in Dahl-S and Dahl-R rats,22 excluding the possibility of autonomic aldosterone secretion in these models. Interestingly, however, previous data indicate that serum- and glucocorticoid-induced kinase 1 (SGK1) and ENaC, the main downstream targets of aldosterone-MR signaling, are paradoxically elevated in salt-loaded Dahl-S rats,34 despite suppression of aldosterone. Consistent with this, we observed increased SGK1 protein and nuclear MR contents in salt-loaded Dahl-S rats. Furthermore, we also found that the selective MR antagonist eplerenone ameliorated high blood pressure and glomerular injury in this model.22 In contrast, salt-loaded Dahl-R rats showed decreased nuclear MR and SGK1 along with reduced plasma aldosterone, indicating that MR signaling is appropriately reduced.
In our previous work, we evaluated the contribution of Rac1 GTPase to the mechanism of this paradoxical MR signaling in salt-loaded Dahl-S rats.22 Interestingly, high salt loading increased Rac1 activity in the kidneys of Dahl-S rats. In contrast, renal Rac1 was reduced by high salt in Dahl-R rats. Pharmacological manipulation of Rac1 by Nsc23766 in Dahl-S rats abrogated hypertension and kidney injury along with reduction in nuclear MR and SGK1 contents, demonstrating the pathological roles of Rac1 overactivity. Thus, these data indicate that salt loading increases Rac1 activity, which in turn causes salt-dependent hypertension via facilitating MR signaling even when circulating aldosterone is reduced (Figure 1).

Rac1 response to high salt loading and salt sensitivity. In salt-resistant phenotype, salt loading decreases plasma aldosterone and also renal Rac1 activity. This results in reduced mineralocorticoid receptor (MR) signaling and natriuresis. In contrast, a subgroup of salt-sensitive hypertension shows increased Rac1 activity by salt loading. The aberrant Rac1 signaling causes MR overactivity in the presence of aldosterone.
Notably, adrenalectomy in Dahl-S rats prevented Rac1 activation, which was restored by exogenous aldosterone supplementation. These data indicate that the Rac1-MR and aldosterone-MR axes are interdependent. They also exclude the possibility that MR is activated by ligands other than aldosterone in this model. Thus, high salt loading suppresses aldosterone but activates MR signaling via Rac1 in the context of salt-sensitive hypertension, contributing to blood pressure elevation and kidney disease progression. Our data indicate that, besides hyperaldosteronism, dysregulation of MR significantly contributes to the pathogenesis of hypertension. Similarly, MR blockade can confer organ protection in kidney diseases associated with pathological MR activity, highlighting the potential usefulness of MR antagonists.
The first-line treatment for proteinuric CKD is angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs).35 Large-scale interventional studies have established that inhibitors of the renin-angiotensin system reduce proteinuria and block CKD progression.36, 37, 38, 39 Importantly, however, the renoprotective effects of ACEIs and ARBs are less clear in patients with high salt intake than in those with low to normal salt intake.40 Given our experimental data, high salt status can be associated with enhanced MR signaling via Rac1, which may not be effectively abrogated by ACEIs or ARBs.
The EVALUATE (Eplerenone Combination vs. Conventional Agents to Lower Blood Pressure on Urinary Anti-albuminuric Treatment Effect) study investigated the effects of long-term, low-dose eplerenone (50 mg per day) added to ACEIs, ARBs or both, on the urinary albumin-to-creatinine ratio in hypertensive patients with nondiabetic CKD (with an estimated glomerular filtration rate of at least 50 ml min−1 1.73 m−2).14 In this randomized, placebo-controlled trial, the authors showed that eplerenone significantly reduced albuminuria and blood pressure in patients with nondiabetic CKD who were already on ACEIs or ARBs. Although serum potassium concentrations were modestly higher in the eplerenone group than the placebo group, none of the participants had hyperkalemia as defined by serum K+ levels higher than 5.5 mmol l−1, indicating that low-dose eplerenone was tolerable in this population. Most interestingly, in post hoc analyses stratified by urinary sodium excretion, the renoprotective effects of eplerenone were prominent in those with high urinary sodium excretion (160 mmol per day or greater). In contrast, there were no significant differences in the percent change in urinary albumin-to-creatinine ratio from baseline between the eplerenone group and placebo group in participants with low urinary sodium excretion (< 160 mmol per day). Furthermore, there was no significant correlation between the percent decrease in urinary albumin-to-creatinine ratio and baseline aldosterone levels. These data indicate the strong association between salt intake and MR signaling in humans, and are consistent with the experimental data showing that salt loading enhances MR signaling without increasing circulating aldosterone. Also, this study clearly demonstrates that MR antagonists have different property to ACEIs or ARBs, and that their use on top of ACEI or ARB may be beneficial at least in a subgroup of CKD patients as far as serum K+ levels are appropriately controlled.
Cell-selective regulation of MR by phosphorylation
In the principal cells of the connecting tubules and collecting duct, aldosterone directly binds to MR and regulates the expression and activity of the amiloride-sensitive ENaC.41 This process is mediated by the interplay of the Ser/Thr kinase SGK1 and ubiquitin ligase neuronal precursor cell expressed developmentally downregulated 4-2.41 Aldosterone regulates ENaC also via proteolytic cleavage42 and epigenetic modification.43 Activation of ENaC by aldosterone drives electrogenic Na+ reabsorption, which promotes lumen-negative potential and stimulates Cl− reabsorption, or K+ and H+ secretion. Consistent with this, in patients with autosomal recessive pseudohypoaldosteronism type I, mutations in any of the three different ENaC subunits cause salt wasting, hyperkalemia and metabolic acidosis.44, 45 K+ secretion in this segment is mediated by renal outer medullary K+ channels, and apical vacuolar H+-ATPase is responsible for H+ secretion.
With regard to the route of Cl− reabsorption in this segment, accumulating data indicate the critical role of pendrin (encoded by SLC26A4), a Cl−/HCO3− exchanger selectively present in β-intercalated cells.46, 47, 48 In mice lacking pendrin, Wall et al.48 found that Cl− flux in the cortical collecting duct is no longer evident, and these mice show the salt-loss phenotype. Conversely, overexpression of pendrin produces salt-dependent hypertension.49 The role of pendrin in the complex network of the kidney fluid system was established in the study by Soleimani et al.,50 who reported severe volume depletion and hypotension in mice lacking pendrin and the Na+–Cl− co-transporter. These data demonstrate that pendrin and Na+–Cl− co-transporter are the main Cl− reabsorption pathways in the distal nephron and that pendrin contributes to fluid and electrolyte homeostasis.
Previous studies have identified the factors influencing pendrin expression and/or activity, including acid/base change, angiotensin II, K+, Cl− and mineralocorticoids.51, 52, 53, 54 Indeed, evidence indicates that intercalated cells express MR.7 In β-intercalated cells, the synthetic mineralocorticoid deoxycorticosterone pivalate is shown to increase pendrin expression in the apical membrane.52 However, the effects of mineralocorticoids may not be evident depending on the experimental conditions,55 suggesting that MR is regulated via complex mechanisms in these cells.
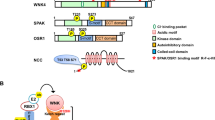
To obtain insights into the mechanisms regulating MR function, we have comprehensively analyzed the phosphorylation sites in MR.25 Among the identified sites, phosphorylation at S843 (MRS843-P) is of particular interest because of its location in the ligand-binding domain. Previous reports have highlighted the importance of S843 in determining ligand selectivity.56 Indeed, biochemical assays using phosphomimetic MRS843E revealed that phosphorylation severely impairs aldosterone binding. Consistent with this, phosphomimic MR is exclusively cytoplasmic and shows no transcriptional activity, even in the presence of aldosterone at a physiological concentration (1 nmol l−1). A recent study suggested that phosphorylated MR exerts a dominant negative effect,57 further underscoring the biological impact of MRS843-P.
In terms of physiological significance, analysis of mouse tissues using phospho-specific antibodies revealed that MRS843-P is exclusively present in the cytoplasm of the intercalated cells and is dephosphorylated in volume depletion via angiotensin II signaling. Conversely, K+ loading increases MRS843-P in these cells. The decrease in MRS843-P in hypovolemic conditions is associated with the increase in pendrin and apical H+-ATPase, which can be corrected by the MR antagonist spironolactone. These data indicate that volume depletion induces MR dephosphorylation, thereby activating Cl− flux mechanisms involving intercalated cells (Figure 2).

Mechanism for mineralocorticoid receptor (MR) regulation in renal intercalated cells. High K+ intake increases MR phosphorylation, which prevents ligand binding and MR activation in hyperaldosteronism. Dephosphorylation of the receptor in response to angiotensin II signaling restores receptor competence, increasing Cl− flux mechanisms involving these cells in the collecting duct. ‘P’ denotes phosphorylation. Adapted from ref. 25.
As elegantly demonstrated by a mathematical model in the previous reports,58, 59 maximal Na–Cl reabsorption in the collecting duct occurs when electrolyte flux mechanisms in intercalated cells and principal cells are activated simultaneously. Under these conditions, K+ secretion is minimized as a result of electroneutral Na–Cl transport in this segment, limiting K+ secretion in the presence of high aldosterone levels. Identification of intercalated cell MR phosphorylation provides a mechanism that integrates the function of principal cells and intercalated cells, which determines the balance of Na+/K+ exchange and Na–Cl co-transport in the collecting duct. Interestingly, we found reduced MRS843-P in a mouse model of pseudohypoaldosteronism type II, a rare Mendelian disorder featuring hypertension and hyperkalemia,25 indicating that the Ser/Thr kinase WNK4 is involved in the regulation of this site. These data also suggest a possibility that the dysregulation of this site may have a role in its pathogenesis. It is likely that pseudohypoaldosteronism type II mice exhibit impaired K+ secretion partly because of the constitutive dephosphorylation of this site.
Future research directions
Identification of a novel MR phosphorylation site raises several questions for further research. First, additional studies are required to determine the identity of the kinase that mediates MRS843-P. Given that phosphorylation at this site non-competitively inhibits ligand binding, artificial manipulation of this site may have therapeutic potential. Second, the mechanisms that control MRS843 phosphorylation in a cell-selective manner are unclear. One possibility is that the responsible kinases are highly enriched in intercalated cells. Alternatively, other post-translational modifications, such as O-linked β-N-acetylglucosamine,60, 61 may have a role. Third, the role of cortisol in intercalated cells may require further investigation. Although the role of aldosterone in intercalated cells has been demonstrated, it is possible that cortisol also acts as a ligand. As suggested previously, the difference in phenotype between MR-knockout mice and aldosterone synthase-knockout mice is consistent with the role of cortisol.62 Finally, the role of MRS843-P in the pathophysiology of hypertension and kidney disease needs further evaluation. Future studies are warranted to clarify these issues, which will hopefully lead to the identification of potential therapeutic targets for selective MR modulation.
References
Hall JE, Guyton AC, Trippodo NC, Lohmeier TE, McCaa RE, Cowley AW Jr . Intrarenal control of electrolyte excretion by angiotensin II. Am J Physiol 1977; 232: F538–F544.
Guyton AC . Blood pressure control—special role of the kidneys and body fluids. Science 1991; 252: 1813–1816.
Lifton RP, Gharavi AG, Geller DS . Molecular mechanisms of human hypertension. Cell 2001; 104: 545–556.
Bakris GL, Williams M, Dworkin L, Elliott WJ, Epstein M, Toto R, Tuttle K, Douglas J, Hsueh W, Sowers J . Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36: 646–661.
Yamagata K, Ishida K, Sairenchi T, Takahashi H, Ohba S, Shiigai T, Narita M, Koyama A . Risk factors for chronic kidney disease in a community-based population: a 10-year follow-up study. Kidney Int 2007; 71: 159–166.
Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM . Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 1987; 237: 268–275.
Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J . In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 2010; 299: F1473–F1485.
Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE . Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 2015; 10: 135–146.
Roy A, Al-bataineh MM, Pastor-Soler NM . Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 2015; 10: 305–324.
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J . The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341: 709–717.
Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M, Eplerenone Post-Acute Myocardial Infarction Heart Failure, E. Survival Study I. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309–1321.
Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B . Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11–21.
Shibata S, Fujita T . Mineralocorticoid receptors in the pathophysiology of chronic kidney diseases and the metabolic syndrome. Mol Cell Endocrinol 2012; 350: 273–280.
Ando K, Ohtsu H, Uchida S, Kaname S, Arakawa Y, Fujita T, Group ES . Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: a double-blind, randomised, placebo-controlled trial. Lancet Diabet Endocrinol 2014; 2: 944–953.
Sato A . The necessity and effectiveness of mineralocorticoid receptor antagonist in the treatment of diabetic nephropathy. Hypertens Res 2015; 38: 367–374.
Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, Remuzzi G, Rossing P, Schmieder RE, Nowack C, Kolkhof P, Joseph A, Pieper A, Kimmeskamp-Kirschbaum N, Ruilope LM, Mineralocorticoid Receptor Antagonist Tolerability Study-Diabetic Nephropathy Study Group. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA 2015; 314: 884–894.
Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T . Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension 2007; 49: 355–364.
Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T . Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med 2008; 14: 1370–1376.
Karashima S, Yoneda T, Kometani M, Ohe M, Mori S, Sawamura T, Furukawa K, Seta T, Yamagishi M, Takeda Y . Comparison of eplerenone and spironolactone for the treatment of primary aldosteronism. Hypertens Res 2016; 39: 133–137.
Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, Lumeng CN, Mortensen RM . Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 2010; 120: 3350–3364.
Nguyen Dinh Cat A, Briones AM, Callera GE, Yogi A, He Y, Montezano AC, Touyz RM . Adipocyte-derived factors regulate vascular smooth muscle cells through mineralocorticoid and glucocorticoid receptors. Hypertension 2011; 58: 479–488.
Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, Kawarazaki W, Takeuchi M, Ayuzawa N, Miyoshi J, Takai Y, Ishikawa A, Shimosawa T, Ando K, Nagase M, Fujita T . Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest 2011; 121: 3233–3243.
Jaffe IZ, Jaisser F . Endothelial cell mineralocorticoid receptors: turning cardiovascular risk factors into cardiovascular dysfunction. Hypertension 2014; 63: 915–917.
Urbanet R, Nguyen Dinh Cat A, Feraco A, Venteclef N, El Mogrhabi S, Sierra-Ramos C, Alvarez de la Rosa D, Adler GK, Quilliot D, Rossignol P, Fallo F, Touyz RM, Jaisser F . Adipocyte mineralocorticoid receptor activation leads to metabolic syndrome and induction of prostaglandin D2 synthase. Hypertension 2015; 66: 149–157.
Shibata S, Rinehart J, Zhang J, Moeckel G, Castaneda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP . Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 2013; 18: 660–671.
Takai Y, Sasaki T, Matozaki T . Small GTP-binding proteins. Physiol Rev 2001; 81: 153–208.
Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, Yoshida H, Doi T, Mizoguchi A, Matsuura N, Niho Y, Nishimune Y, Nishikawa S, Takai Y . Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene 1999; 18: 5373–5380.
Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, Hatori T, Tsuchiya A, Kiyono M, Nosaka T, Nakajima H, Williams DA, Kitamura T . Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J Cell Biol 2006; 175: 937–946.
Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F . Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 2008; 133: 340–353.
Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, Beck BB, Gribouval O, Zhou W, Diaz KA, Natarajan S, Wiggins RC, Lovric S, Chernin G, Schoeb DS, Ovunc B, Frishberg Y, Soliman NA, Fathy HM, Goebel H, Hoefele J, Weber LT, Innis JW, Faul C, Han Z, Washburn J, Antignac C, Levy S, Otto EA, Hildebrandt F . ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 2013; 123: 3243–3253.
Gupta IR, Baldwin C, Auguste D, Ha KC, El Andalousi J, Fahiminiya S, Bitzan M, Bernard C, Akbari MR, Narod SA, Rosenblatt DS, Majewski J, Takano T . ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet 2013; 50: 330–338.
Fujita T, Henry WL, Bartter FC, Lake CR, Delea CS . Factors influencing blood pressure in salt-sensitive patients with hypertension. Am J Med 1980; 69: 334–344.
Rapp JP, Dahl LK . Suppression of aldosterone in salt susceptible and salt resistant rats. Endocrinology 1973; 92: 1286–1289.
Farjah M, Roxas BP, Geenen DL, Danziger RS . Dietary salt regulates renal SGK1 abundance: relevance to salt sensitivity in the Dahl rat. Hypertension 2003; 41: 874–878.
James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS, Ortiz E . 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311: 507–520.
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD . The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329: 1456–1462.
Ruggenenti P, Perna A, Gherardi G, Gaspari F, Benini R, Remuzzi G . Renal function and requirement for dialysis in chronic nephropathy patients on long-term ramipril: REIN follow-up trial. Gruppo Italiano di Studi Epidemiologici in Nefrologia (GISEN). Ramipril Efficacy in Nephropathy. Lancet 1998; 352: 1252–1256.
Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345: 851–860.
Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, Investigators RS . Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345: 861–869.
Vegter S, Perna A, Postma MJ, Navis G, Remuzzi G, Ruggenenti P . Sodium intake, ACE inhibition, and progression to ESRD. J Am Soc Nephrol 2012; 23: 165–173.
Pearce D, Kleyman TR . Salt, sodium channels, and SGK1. J Clin Invest 2007; 117: 592–595.
Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, Shiraishi N, Nonoguchi H, Chen LM, Chai KX, Chao J, Tomita K . Regulation of prostasin by aldosterone in the kidney. J Clin Invest 2002; 109: 401–408.
Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC . Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 2007; 117: 773–783.
Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP . Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet 1996; 12: 248–253.
Strautnieks SS, Thompson RJ, Gardiner RM, Chung E . A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet 1996; 13: 248–250.
Schlatter E, Greger R, Schafer JA . Principal cells of cortical collecting ducts of the rat are not a route of transepithelial Cl- transport. Pflugers Arch 1990; 417: 317–323.
Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED . Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 2001; 98: 4221–4226.
Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW . NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension 2004; 44: 982–987.
Jacques T, Picard N, Miller RL, Riemondy KA, Houillier P, Sohet F, Ramakrishnan SK, Busst CJ, Jayat M, Corniere N, Hassan H, Aronson PS, Hennings JC, Hubner CA, Nelson RD, Chambrey R, Eladari D . Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol 2013; 24: 1104–1113.
Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, Amlal H . Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA 2012; 109: 13368–13373.
Frische S, Kwon TH, Frokiaer J, Madsen KM, Nielsen S . Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. Am J Physiol Renal Physiol 2003; 284: F584–F593.
Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM . Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 2003; 42: 356–362.
Hafner P, Grimaldi R, Capuano P, Capasso G, Wagner CA . Pendrin in the mouse kidney is primarily regulated by Cl- excretion but also by systemic metabolic acidosis. Am J Physiol Cell Physiol 2008; 295: C1658–C1667.
Verlander JW, Hong S, Pech V, Bailey JL, Agazatian D, Matthews SW, Coffman TM, Le T, Inagami T, Whitehill FM, Weiner ID, Farley DB, Kim YH, Wall SM . Angiotensin II acts through the angiotensin 1a receptor to upregulate pendrin. Am J Physiol Renal Physiol 2011; 301: F1314–F1325.
Mohebbi N, Perna A, van der Wijst J, Becker HM, Capasso G, Wagner CA . Regulation of two renal chloride transporters, AE1 and pendrin, by electrolytes and aldosterone. PLoS ONE 2013; 8: e55286.
Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW . Crystal structure of an ancient protein: evolution by conformational epistasis. Science 2007; 317: 1544–1548.
Jimenez-Canino R, Fernandes MX, Alvarez de la Rosa D . Phosphorylation of mineralocorticoid receptor ligand binding domain impairs receptor activation and has a dominant negative effect over non-phosphorylated receptors. J Biol Chem 2016; 291: 19068–19078.
Weinstein AM . A mathematical model of rat collecting duct. III. Paradigms for distal acidification defects. Am J Physiol Renal Physiol 2002; 283: F1267–F1280.
Wall SM, Weinstein AM . Cortical distal nephron Cl(-) transport in volume homeostasis and blood pressure regulation. Am J Physiol Renal Physiol 2013; 305: F427–F438.
Butkinaree C, Park K, Hart GW . O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta 2010; 1800: 96–106.
Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O . Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 2011; 80: 825–858.
Funder JW . Angiotensin retains sodium by dephosphorylating mineralocorticoid receptors in renal intercalated cells. Cell Metab 2013; 18: 609–610.
Acknowledgements
This work was supported in part by JSPS KAKENHI grants 15H04837 (to S.S.); Banyu Life Science Foundation International (to S.S.); Kanae Foundation for the Promotion of Medical Science (to S.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
This work was supported in part by Banyu Life Science Foundation International given to S.S.
Rights and permissions
About this article

Cite this article
Shibata, S., Ishizawa, K. & Uchida, S. Mineralocorticoid receptor as a therapeutic target in chronic kidney disease and hypertension. Hypertens Res 40, 221–225 (2017). https://doi.org/10.1038/hr.2016.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2016.137
Keywords
This article is cited by
-
Prediction of endogenous mineralocorticoid receptor activity by depressor effects of mineralocorticoid receptor antagonists in patients with primary aldosteronism
Hypertension Research (2024)
-
Fluid homeostasis induced by sodium-glucose cotransporter 2 inhibitors: novel insight for better cardio-renal outcomes in chronic kidney disease
Hypertension Research (2023)
-
Esaxerenone for nocturnal hypertension and possible future direction for treatment of hypertension-cardiovascular-kidney comorbidity
Hypertension Research (2023)
-
Ameliorative effect of mussel-derived ACE inhibitory peptides on spontaneous hypertension rats
European Journal of Nutrition (2023)
-
Efficacy and safety of a low-sodium diet and spironolactone in patients with stage 1-3a chronic kidney disease: a pilot study
BMC Nephrology (2022)
