
Abstract
Quantitative genetics approaches, and particularly animal models, are widely used to assess the genetic (co)variance of key fitness related traits and infer adaptive potential of wild populations. Despite the importance of precision and accuracy of genetic variance estimates and their potential sensitivity to various ecological and population specific factors, their reliability is rarely tested explicitly. Here, we used simulations and empirical data collected from an 11-year study on tree swallow (Tachycineta bicolor), a species showing a high rate of extra-pair paternity and a low recruitment rate, to assess the importance of identity errors, structure and size of the pedigree on quantitative genetic estimates in our dataset. Our simulations revealed an important lack of precision in heritability and genetic-correlation estimates for most traits, a low power to detect significant effects and important identifiability problems. We also observed a large bias in heritability estimates when using the social pedigree instead of the genetic one (deflated heritabilities) or when not accounting for an important cause of resemblance among individuals (for example, permanent environment or brood effect) in model parameterizations for some traits (inflated heritabilities). We discuss the causes underlying the low reliability observed here and why they are also likely to occur in other study systems. Altogether, our results re-emphasize the difficulties of generalizing quantitative genetic estimates reliably from one study system to another and the importance of reporting simulation analyses to evaluate these important issues.
Similar content being viewed by others
Introduction
Understanding the genetic component underlying important fitness related traits is essential to infer the adaptive potential of wild populations (Lynch and Walsh, 1998; Hendry et al., 2011). However, as most traits are likely under the control of numerous genes with small effects (Lande, 1981; Falconer and Mackay, 1996; Husby et al., 2015), potentially interacting with the genetic components of other traits (Lande and Arnold, 1983; Blows and Hoffmann, 2005) and/or with the environment in which they are expressed (Via and Lande, 1985; Hoffmann and Merilä, 1999), this task remains challenging despite rapid advances in whole-genome analysis methods (Vinkhuyzen et al., 2013). For these reasons, quantitative genetic approaches—statistical methods using known relationships between individuals to assess the genetic and environmental components of a population phenotypic variance—remain efficient ways to infer the underlying genetic variation of focal traits (Falconer and Mackay, 1996; Kruuk et al., 2008). For this purpose, animal models are particularly suitable for wild population datasets because they allow the use of all relationships between individuals and can account for missing data, unbalanced designs and other potential biases (for example, environmental causes of phenotypic similarity among individuals rather than genetic ones) (Henderson, 1984; Kruuk, 2004; Kruuk and Hadfield, 2007; Wilson et al., 2010). Moreover, heritability estimates from wild populations obtained with animal models are generally more accurate than those estimated with traditional parent–offspring regressions (reviewed in Postma, 2014).
Several factors will influence the validity of the genetic variance estimates obtained in the wild, which in turn will affect our capacity to infer population responses to selective pressures (for example, through breeder’s or Robertson–Price equation, see Morrissey et al., 2010). Precision—the repeatability of a measurement—is highly dependent on sample size both in terms of pedigree depth and completeness, but also on the complexity of the genetic architecture underlying the focal trait (Morrissey et al., 2007; de Villemereuil et al., 2013). For example, given two pedigrees with identical sample sizes, a pedigree with a weak degree of connectivity between individuals, for example, in populations with strong natal dispersion and/or immigration resulting in few related individuals, will be less precise than a pedigree with known relationships between all individuals (Wilson et al., 2010). However, precision is rarely tested per se, but rather deduced from the size of standard errors around estimates (for example, Charmantier and Réale, 2005). Accuracy—the proximity between the estimated value and the true value—is affected mainly by pedigree errors (that is, erroneous links between individuals in the pedigree; Charmantier and Réale, 2005; Morrissey et al., 2007; Firth et al., 2015) and by model parameterizations (Kruuk and Hadfield, 2007; Wilson, 2008; Wolak et al., 2015). Pedigree errors could be particularly common in study systems where extra-pair copulations are important and parental links in pedigrees constructed solely based on social pair observations (that is, social pedigree). Theoretically, the presence of extra-pair paternity (EPP), if not accounted for, can bias downward the heritability estimates, even though evidence so far suggests that this bias is generally small in wild populations (Charmantier and Réale, 2005; Bérénos et al., 2014; Firth et al., 2015; but see Lee and Pollak, 1997 for reports of bias in the animal breeding litterature; see also Walther and Moore, 2005 for a detailed discussion on the concepts of bias, precision and accuracy).
Despite previous knowledge of what can affect precision and accuracy of genetic variance estimates, extrapolating the reliability of these estimates from one study system to another is difficult given the large diversity of life history observed across species that inevitably modified dataset and pedigree structures. Therefore, simulation analyses are recommended for testing the limits of a particular dataset, pedigree and model to answer specific biological questions (Morrissey et al., 2007; Wilson et al., 2010). Simulation frameworks allow the assessment of power—the probability of detecting an effect given that this effect is true—of a particular dataset or model. In animal models, power is affected by the same factors as precision (Quinn et al., 2006; Morrissey et al., 2007; Bérénos et al., 2014). However, performing a power analysis alone could provide an incomplete picture of the validity of genetic variance estimates. In fact, a particular model applied on a dataset could have a high power to estimate a given effect, but the model itself might be missing a critical variance term, leading to precise but inaccurate estimates (Kruuk and Hadfield, 2007). Similarly, even if all important variance terms are included in a particular model, the model variance structure might not allow discrimination between two variance terms being included, a problem of statistical models related to their low ‘identifiability’ (Bolker, 2008). Confounded parameters could be frequent when applying animal models to wild populations (Wilson et al., 2010), but the extent of identifiability problems is still unknown. Finally, reliability of quantitative genetics estimates is even less reported for multivariate models than univariate models, limiting our confidence on important parameter estimations such as genetic additive covariance/correlation among traits.
Here, we used an 11-year study on Tree swallow (Tachycineta bicolor (Vieillot, 1808)) to assess the effects of identity errors, structure and size of a pedigree on reliability of quantitative genetic estimates. Tree swallows are small migratory passerines and their breeding grounds are widely distributed across North America (Winkler et al., 2011). Similarly to other migrating species, tree swallow shows high natal dispersal, high mortality within the first year and low nestling recruitment rate (Hosner and Winkler, 2007), which reduce kinship among individuals in monitored populations. This socially monogamous species also displays one of the highest rate of EPP documented in birds (Griffith et al., 2002), with more than 80% of nests containing at least one extra-pair offspring and overall around 50% of nestlings resulting from extra-pair copulations (Dunn et al., 2001; Lessard et al., 2014). In the context of building a pedigree for quantitative genetics analyses, this means that half of paternal links would be erroneous if using a social pedigree. This represents a higher proportion of EPP than those tested in previous studies to assess their impacts on heritability estimates (for example, in Charmantier and Réale, 2005; Firth et al., 2015), but it is a proportion that will be typically found in other similar study systems (see Griffith et al., 2002 and references therein). Furthermore, reliability of genetic variance estimates for this and similar species is difficult to predict based on previous results obtained on other species, as the typical poorly connected pedigree available for migratory species could be compensated by the natural half-sibs design resulting from the high rate of EPP occurring in tree swallows.
In this study, we used a social and a genetic pedigree to estimate and compare genetic additive (co-)variances and heritability of traits differing in their completeness through the pedigree structure. More specifically, we first used empirical data to assess the bias resulting from using a social pedigree over a genetic one. Then, we used simulated data to assess precision and accuracy of quantitative genetic estimates obtained with both pedigrees, as well as power of datasets and models, and identifiability among variance terms.
Materials and methods
Study species, system and phenotypic data
Tree swallows breed in tree cavities or nest boxes, they produce only one clutch per year, containing on average five eggs and both parents provide care to nestlings (Winkler et al., 2011). Since 2004, we have intensively followed tree swallow activities during the breeding season at 400 nest boxes equally distributed across 40 farms in an area covering 10 200 km2 in southern Québec, Canada (detailed in Ghilain and Bélisle, 2008). Nest boxes were visited every 2 days from May to July to record nest box occupancy and important brood characteristics (for example, laying date, clutch size and hatching date). Adults and nestlings were individually marked with an aluminum band (US Fish and Wildlife Service). Adults were captured directly in their nest box by a trap system, during the incubation period and food provisioning period for females and males, respectively. Morphological measurements of body mass (±0.01 g), non-flattened wing length (±0.5 mm) and tarsus length (±0.02 mm) were taken on adult tree swallows during captures. Females were classified based on their plumage color as second-year (brown) or after-second-year (blue-green) (Hussell, 1983) and a minimal age was determined for all adults based on the year they were first observed in the study system. Nestlings were captured at 16 days old before they fledged (fledging occurs around 18–22 days) to record body mass (±0.01 g), primary length (hereafter wing length; ±0.02 mm) and tarsus length (±0.02 mm). Since 2006, blood samples of adults and nestlings have been collected on a qualitative P8 grade filter paper (Thermo Fisher Scientific, Waltham, MA, USA) for molecular analysis (see below).
To reflect the large differences that can exist between traits in terms of sample size and amount of standing genetic variance, we focused on nine phenotypic traits grouped in three categories: (1) morphological traits, which included wing length, body mass and tarsus length of all adults; (2) reproductive traits, which were restricted to females, and included laying date (that is, date of the first egg laid), clutch size (that is, number of eggs laid within a nest box) and incubation duration (defined as hatching date−(laying date+clutch size−1)); and (3) nestling traits, which included wing length, body mass and tarsus length measured at the age of 16 days. Most traits were measured since the beginning of the research program (that is, in 2004), but some traits were first measured later (nestling body mass: 2005; nestling wing length: 2006; adult/nestling tarsus length: 2007) creating differences in data completeness among traits (sample size per traits can be found in Supplementary Table A1).
Molecular analysis and pedigree construction
DNA extraction, molecular sexing and microsatellite data analyses are detailed in Lessard et al. (2014). Briefly, DNA was extracted from blood samples following a standard salt-extraction method and DNA concentration was determined by migration on 2% agarose gels with a molecular weight standard. A molecular sexing technique was used to determine nestling sexes and to validate adult field observations. All DNA samples were characterized at six microsatellites loci using an AB3130xl automated DNA sequencer and allele lengths were determined using GeneMapper v4.1 (Applied Biosystems, Foster City, CA, USA).
We constructed a social pedigree using social male identities (that is, males caught in nest boxes while feeding the young) and a genetic pedigree using genetic father identities (that is, males assigned as fathers using genetic analyses—see below). In both pedigrees, dam identities were first determined by female captures during egg incubation and then verified genetically based on locus mismatches with nestlings (2.1% of broods with two females captured within the same nest box, from which only 3.6% were from mixed maternity; 11 nestling genotypes (0.15%) mismatched at more than two loci with their social mother genotype). Genetic fathers were determined by parental assignations with CERVUS v3.0.7 (Kalinowski et al., 2007; Lemons et al., 2015) in a three step procedure. First, we proceeded to father assignments each year separately following a method slightly modified from Lessard et al. (2014). Candidate fathers considered in analyses included all males captured during a given breeding season within 15 km of the nest box of interest (see Lessard et al., 2014 for the rationale behind this approach), but also all males not captured in a given year but suspected of being present outside of our nest box system (that is, captured on the same farm on both previous and following years). These assignations were based on a 90% confidence level, assuming a 2% mistyping error rate and we used the percentage of social fathers captured as the percentage of candidate fathers known (variable among years, range: 64–88%). Second, social fathers, when known, were tested for being genetic fathers of offspring in their nest using the likelihood-based approach of Lemons et al. (2015). Briefly, we re-ran the parental analysis with the social father as the unique candidate father for a given nestling, and we defined the proportion of sampled fathers as the proportion of nestlings without any locus mismatch with its social father (that is, the probability of the social male being the true father). We then extracted, for each nestling, the critical logarithm of odds score associated with 95% confidence that its social father was not its true father and we compared these scores with those observed in regular parental analyses. Males significantly assigned to nestlings in the initial parental analysis (that is, step 1) were considered as their genetic fathers. For the remaining nestlings (that is, without a significant male assignment at step 1), if their social fathers could not be excluded (that is, step 2), they were considered as their genetic fathers but otherwise no genetic father were assigned to them (see Supplementary Figure A1 for the exact number of fathers assigned to a nestling at each step).
Summary statistics for both pedigrees were obtained with the package PEDANTICS (Morrissey et al., 2007; Morrissey and Wilson, 2010) in R v3.2.0 (R Core Team, 2015). These statistics were computed for complete pedigrees, but also for pedigrees pruned to contain only informative individuals based on the availability of phenotypic data for each trait (hereafter pruned pedigrees) and are presented for each trait category in Table 1 (see also Supplementary Table A2 for more information on each trait).
Estimations of quantitative genetic parameters
To estimate additive genetic (co)variances of our focal traits, we used both univariate and multivariate animal models (Kruuk, 2004; Wilson et al., 2010). Fixed effects (for example, age, sex) were included for some traits based on mixed model analyses detailed in Supplementary Information. For adult morphological and reproductive traits, full univariate animal models were constructed as follows:

and for nestling traits:

where VP is the phenotypic variance after accounting for fixed effects, VA is the additive genetic variance, VPE is the permanent environmental effect, VY and VBY are the variance among years and among birth years, respectively, VB is the variance among broods and VR the residual variance. All traits followed a Gaussian distribution and animal models were resolved with a restricted maximum likelihood method (REML), using both the social and the genetic pedigree. Final animal models were constructed by sequential model-building from a model with only residual variance to more complex ones, with a comparison of models at each step using a likelihood ratio test (LRT; see Supplementary Tables C1 for the increasing levels of complexity). Only VA in the incubation duration models did not significantly improve the model likelihood (χ2<0.01, P>0.99).
We also constructed three multivariate animal models, one for each trait category. For each of them, we first included the same variance terms as for univariate models. However, due to convergence problems when including VPE for morphological and reproductive traits, we decided to use a model without the VPE term on a reduced dataset comprising only one observation by individual (randomly chosen). Moreover, as we observed no VA for incubation duration, this trait was not included in the multivariate analysis of reproductive traits. Covariances among traits for each variance components were estimated using unstructured variance models. Significance was tested by comparing a model including covariance estimation to a model where covariance was constrained to be equal to zero using LRT.
We estimated heritability (h2=VA/VP, Falconer and Mackay, 1996) and coefficient of genetic variation (CVA= , where
, where  is the trait mean, Houle, 1992) for all traits within each analysis. For multivariate analyses we also estimated additive genetic correlations (rA) between each pair of traits. All animal model analyses were conducted with ASRemL v3.0.5 (VSN International Ltd, Hemel Hempstead, UK). Standard errors (SEs) for variance components and h2 estimates were computed directly by ASRemL. For CVA, standard deviations were approximated following the equation provided by Lynch and Walsh (1998; equation A1.26c, Appendix 1).
is the trait mean, Houle, 1992) for all traits within each analysis. For multivariate analyses we also estimated additive genetic correlations (rA) between each pair of traits. All animal model analyses were conducted with ASRemL v3.0.5 (VSN International Ltd, Hemel Hempstead, UK). Standard errors (SEs) for variance components and h2 estimates were computed directly by ASRemL. For CVA, standard deviations were approximated following the equation provided by Lynch and Walsh (1998; equation A1.26c, Appendix 1).
Simulation analyses
We simulated three different datasets of phenotypic data within both the social and genetic pedigree structures with different levels of complexity. In all cases, simulated traits were normally distributed among all individuals, with VP=1,  =0 and h2 of 0.1, 0.3 and 0.5 for the focal trait. In dataset 1, we simulated one phenotype per individual with the parameters mentioned above to reflect the simplest scenario possible. In dataset 2, we simulated phenotypic traits with a more complex underlying structure based on equations (1) and (2) to better reflect our empirical dataset, with similar value of h2 as dataset 1. For nestling traits, we simulated phenotypes with VY=0.1 and VB=0.3, whereas for morphological and reproductive traits we simulated phenotypic values with VY=0.1 and VPE=0.1, thus implying multiple observations per individual. Finally, in dataset 3, different genetic and environmental correlations between traits were also implemented in simulations to assess the difference in power, precision and accuracy when using multivariate models. More specifically, we simulated phenotypes with h2 similar to dataset 1 for the focal trait, whereas fixing h2 of the two other traits at 0.3, for three different values of rA, 0.1, 0.3 and 0.5, whereas fixing environmental correlation at 0.3. No other variance components were simulated. Simulations were performed in R, and breeding values were simulated with the package PEDANTICS. Although simulations were performed within total social and genetic pedigree structures, only individuals with complete information in the empirical dataset were kept in these three simulated datasets.
=0 and h2 of 0.1, 0.3 and 0.5 for the focal trait. In dataset 1, we simulated one phenotype per individual with the parameters mentioned above to reflect the simplest scenario possible. In dataset 2, we simulated phenotypic traits with a more complex underlying structure based on equations (1) and (2) to better reflect our empirical dataset, with similar value of h2 as dataset 1. For nestling traits, we simulated phenotypes with VY=0.1 and VB=0.3, whereas for morphological and reproductive traits we simulated phenotypic values with VY=0.1 and VPE=0.1, thus implying multiple observations per individual. Finally, in dataset 3, different genetic and environmental correlations between traits were also implemented in simulations to assess the difference in power, precision and accuracy when using multivariate models. More specifically, we simulated phenotypes with h2 similar to dataset 1 for the focal trait, whereas fixing h2 of the two other traits at 0.3, for three different values of rA, 0.1, 0.3 and 0.5, whereas fixing environmental correlation at 0.3. No other variance components were simulated. Simulations were performed in R, and breeding values were simulated with the package PEDANTICS. Although simulations were performed within total social and genetic pedigree structures, only individuals with complete information in the empirical dataset were kept in these three simulated datasets.
Different analyses were performed on simulated datasets to answer two different questions. First, to assess if there was a bias created by not accounting for EPP, we used datasets simulated with the genetic pedigree in animal models using either the genetic pedigree (GG analysis) or the social pedigree (GS analysis), and then the results were compared. Second, to look at inherent differences in reliability caused by pedigree structures, datasets simulated with the social pedigree were analyzed using the social pedigree (SS analysis) and were compared with GG analysis. Animal models used for datasets 1 and 3 included only VA and VR as variance components (and covariance in dataset 3), whereas for dataset 2 they included all components described in equations (1) and (2). All these scenarios within each analysis were repeated 100 times for each trait, and animal model analyses were conducted with ASRemL.
Precision and accuracy for h2 and rA estimates were checked visually with boxplots (that is, median for accuracy and distribution of estimates for precision) and also by computing the mean squared error (MSE) for each scenario. MSE is defined as  , where d is the true value (for example, the simulated parameter of h2 or rA; these values are highly correlated (r>0.99) with the realized value within our simulated population) and
, where d is the true value (for example, the simulated parameter of h2 or rA; these values are highly correlated (r>0.99) with the realized value within our simulated population) and  is the estimated value; a small MSE indicates high precision and accuracy (Bolker, 2008). Root MSE (RMSE) was used to allow a comparison at the scale of the estimates (de Villemereuil et al., 2013). Power to detect significant h2 and rA within each scenario were estimated using a ‘rule of thumb’ approach by computing the proportion of estimates that were two times larger than their SE. Although LRTs could have provided a formal hypothesis testing, they were not possible to implement in our simulations procedures. Instead, the ‘rule of thumb’ approach is a practical indicator of statistical testing that could be easily integrated to simulation analyses and lead to results comparable to a LRT approach (Supplementary Table A3; see also Wilson et al., 2011 for a similar approach). Finally, we also checked the identifiability of variance terms in dataset 2 using a Spearman’s rank correlation between estimates of phenotypic variance components. In this case, a low correlation between two components would indicate a high identifiability, whereas a high correlation would indicate a low identifiability.
is the estimated value; a small MSE indicates high precision and accuracy (Bolker, 2008). Root MSE (RMSE) was used to allow a comparison at the scale of the estimates (de Villemereuil et al., 2013). Power to detect significant h2 and rA within each scenario were estimated using a ‘rule of thumb’ approach by computing the proportion of estimates that were two times larger than their SE. Although LRTs could have provided a formal hypothesis testing, they were not possible to implement in our simulations procedures. Instead, the ‘rule of thumb’ approach is a practical indicator of statistical testing that could be easily integrated to simulation analyses and lead to results comparable to a LRT approach (Supplementary Table A3; see also Wilson et al., 2011 for a similar approach). Finally, we also checked the identifiability of variance terms in dataset 2 using a Spearman’s rank correlation between estimates of phenotypic variance components. In this case, a low correlation between two components would indicate a high identifiability, whereas a high correlation would indicate a low identifiability.
Results
Pedigrees
Our total dataset comprised 13 446 individuals, with 2839 individuals observed only as adults, 10 472 observed only as nestlings and 135 observed at both states (that is, recruits). From the field observations, we were able to determine the identity of the social father for 69.2% of nestlings (82.2% since 2006). Parental analyses allowed us to determine the genetic father for 53.3% of nestling (66.3% since 2006). On average, 1.74±0.82 (range: 1−5) genetic fathers were assigned per brood, and each adult male had on average 4.81±5.44 (range: 0−37) nestlings assigned over its entire lifespan. Overall, 49.3% of nestlings successfully genotyped with a known social father were extra-pair young (Ntotal=6382).
Pedigree size varied among the type of pedigree and traits (Table 1). As expected, the social pedigree always had a higher number of links related to father identities than its genetic counterpart, a difference mainly caused by unsampled males contributing to the genetic pool of nestlings. Nestling traits had a much higher number of links than morphological and reproductive traits, which were both of similar order of magnitude, but with slightly fewer links for the latter (Table 1). Pedigree sizes were similar for each trait within a given category (Supplementary Table A2).
Estimations of quantitative genetic parameters (empirical data)
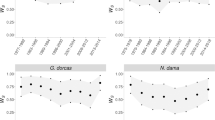
Variance components, and resulting heritability values, estimated from empirical data varied among traits and also often importantly between pedigrees (social versus genetic) and between models (univariate versus multivariate) (Figure 1; Table 2). Biases in h2 and CVA resulting from using social instead of genetic pedigrees were moderate for morphological traits (h2: −0.21 to −0.02; CVA: −0.008 to −0.001), almost null for reproductive traits (almost all around 0) and highly variable for nestling traits (h2: −0.04–0.21; CVA: −0.001–0.051). Estimates of rA obtained with the social and genetic pedigree were similar, except between nestling wing length and body mass where rA was positive and significantly different from 0 with the social pedigree (rA=0.56±0.13, χ2=9.92, P=0.002), but not with the genetic pedigree (rA=0.25±0.19, χ2=1.32, P=0.25; Supplementary Figure C1). Raw phenotypic variance for all traits can be found in Supplementary Table C4.

Proportion of phenotypic variance estimated from final animal models on empirical dataset, for (a) morphological, (b) reproductive and (c) nestling traits. Different models were assessed for each trait: univariate models using social (SU) and genetic (GU) pedigree, and multivariate models using social (SM) and genetic (GM) pedigree.
Reliability of quantitative genetics estimates (simulated data)
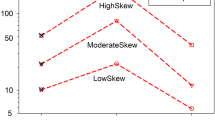
Visual inspection of boxplots (Figures 2b, d, f and 3b) and comparison of RMSE values (Supplementary Figures D1) showed complementary information on precision and accuracy of h2 and rA estimates. Precision of h2 and rA estimates varied greatly depending on trait category (nestling>morphological>reproductive; mean±s.d. RMSE for h2/rA:  =0.055±0.045/0.081±0.025,
=0.055±0.045/0.081±0.025,  =0.122±0.026/0.342±0.061,
=0.122±0.026/0.342±0.061,  =0.200±0.045/0.430±0.025) but also among datasets used (dataset 2 (simulation of h2 and other source of resemblance among individuals)>3 (simulation of h2 and rA)>1 (simulation of h2 only); mean±s.d. RMSE for h2:
=0.200±0.045/0.430±0.025) but also among datasets used (dataset 2 (simulation of h2 and other source of resemblance among individuals)>3 (simulation of h2 and rA)>1 (simulation of h2 only); mean±s.d. RMSE for h2:  =0.144±0.086,
=0.144±0.086,  =0.100±0.047,
=0.100±0.047,  =0.127±0.072). Accuracy for h2 estimates was high for all GG and SS analyses (except for reproductive traits in dataset 3), but downwardly biased in GS analyses for nestling and reproductive traits, an effect more important as simulated h2 increased (mean±s.d. RMSE:
=0.127±0.072). Accuracy for h2 estimates was high for all GG and SS analyses (except for reproductive traits in dataset 3), but downwardly biased in GS analyses for nestling and reproductive traits, an effect more important as simulated h2 increased (mean±s.d. RMSE:  =0.117±0.076,
=0.117±0.076,  =0.143±0.061,
=0.143±0.061,  =0.115±0.075). Accuracy for rA were similar for all type of analysis (mean±s.d. RMSE:
=0.115±0.075). Accuracy for rA were similar for all type of analysis (mean±s.d. RMSE:  =0.283±0.160,
=0.283±0.160,  =0.294±0.151,
=0.294±0.151,  =0.275±0.158).
=0.275±0.158).

Power, precision and accuracy of heritability estimated from animal models for dataset 1 (a and b; simulation of h2 only), dataset 2 (c and d; simulation of h2 and other source of resemblance among individuals) and dataset 3 (e and f; simulation of h2 and rA) simulated on the pedigree structure of morphological traits (dark gray lines), reproductive traits (light gray lines) and nestling traits (black lines). Power of detecting significant heritability over 300 simulated trait values (100 simulations per trait) are presented in a, c and e. Distributions of these estimates are represented by boxplots (1st quartile, median, 3rd quartile) for 2 levels of heritability (dotted lines represent the h2 simulated value of 0.1 and 0.5) in b, d and f. Analysis types refer to datasets simulated with the genetic pedigree and analyzed with the genetic (GG analysis, solid lines) or the social pedigree (GS analysis, dashed lines), and datasets simulated with the social pedigree and analyzed with the social pedigree (SS analysis, dotted lines). For graphical representation, rA was fixed at 0.3 in e and f.

Power, precision and accuracy of genetic additive correlation estimated from multivariate animal models, for dataset 3 (simulation of h2 and rA) simulated on the pedigree structure of morphological traits (dark gray lines), reproductive traits (light gray lines) and nestling traits (black lines). Power of detecting significant genetic correlation over 300 simulated trait values (100 simulations per trait), is presented in a and distributions of these estimates are represented by boxplots (1st quartile, median, 3rd quartile) in b (for 1 value of rA—dotted lines represent the rA true value of 0.5 simulated; 5 estimates>1 are not presented). Analysis types refer to datasets simulated with the genetic pedigree and analyzed with the genetic (GG analysis, solid lines) or the social pedigree (GS analysis, dashed lines), and datasets simulated with the social pedigree and analyzed with the social pedigree (SS analysis, dotted lines). For graphical representation, h2 was fixed at 0.5 in b.
Power to detect significant h2 or rA was greater for traits with larger sample size (nestling>morphological>reproductive; Figures 2a, c, e and 3a), but we observed no difference among traits within each category (data not shown). For multivariate animal models from dataset 3, power to detect h2 was similar for the different genetic correlations tested (data not shown), whereas power to detect genetic correlation increased with increasing heritability (Figure 3a). Within a trait category, power was similar between GG and SS analyses, except for nestling traits from dataset 2 were the simulated brood effect decreased power in SS analysis (Figure 2c). Finally, power comparison between GG and GS analyses revealed generally lower power in morphological and nestling trait for GS (Figures 2a, c, e and 3a).
For adult traits, VA and VPE estimates from dataset 2 were highly negatively correlated, suggesting that these terms were almost completely confounded (morphological traits: rs=−0.95, P<0.001; reproductive traits: rs=−0.94, P<0.001, results from GG analysis with h2=0.3; Supplementary Figures D3). For nestling traits, VA and VB estimates were also correlated, but to a lesser extent (rs=−0.42, P<0.001; Supplementary Figure D5). All other variance terms showed correlations non-significantly different from 0 (all |rs|<0.09, P>0.12).
Finally, given the large differences observed for some reproductive traits between SEs of h2 estimates from empirical and simulated dataset (Table 2; Supplementary Figure D6a), we further inspected the impact of model parameters on SEs. More specifically, we assessed the impact of not accounting for an important cause of resemblance among individuals by running dataset 2 animal models and omitting VPE or VB components. By doing so, we observed a power of 1 at all traits, and all h2 estimates were very precise but showed a bias equivalent to the variance component not accounted for (that is, 0.1 for morphological and reproductive trait and 0.4 for nestling traits; Figure 4). As suspected, SEs for these biased h2 estimates were small and similar to those obtained in empirical analyses, where VPE was almost completely confounded with VA (Supplementary Figure D6b).

Precision and accuracy of heritability estimated from animal models omitting an important cause of resemblance among individuals (VPE or VB), for morphological traits, reproductive traits and nestling traits simulated in dataset 2. Distribution of these estimates are represented by boxplots (1st quartile, median, 3rd quartile) for 2 levels of heritability (dotted lines represent the h2 true value of 0.1 and 0.5 simulated). Analysis types refer to datasets simulated with the genetic pedigree and analyzed with both the genetic and the social pedigree (GG and GS analyses, respectively), and datasets simulated with the social pedigree and analyzed with the social pedigree (SS analysis).
Discussion
Despite the importance of obtaining precise and accurate genetic (co)variance estimates when assessing a population’s adaptive potential, their reliability is rarely explicitly tested or reported in wild populations. Here, we formally assessed the reliability of genetic (co)variance estimates, in a species with a high rate of extra-pair reproduction and low recruitment, with a combination of empirical and simulated data. Altogether, our simulation analyses emphasized the limits of this particular dataset by revealing an important lack of precision in h2 and rA estimates for all adult traits, a lack of power to detect significant effects and identifiability problems between VA and VPE. Moreover, we observed a large bias in h2 when using the social pedigree instead of the genetic one, and also when non-genetic causes of resemblance among individuals (that is, repeated measurements or brood effects) were not accounted for in our analyses. We briefly discuss below (i) the factors contributing to estimate reliability, (ii) the caution needed with model parameterization, (iii) the impacts of high levels of EPP on genetic variance estimates and (iv) we finally conclude with remarks on the accessibility and utility of simulation analyses to address all these potential problems.
Estimate reliability and generalization among studies
At first glance, our sample size seemed large enough to be powerful, with more than 10 years of sampling and a number of records within our pruned pedigrees that was larger than the median number of records typically reported for similar studies in the literature. Indeed, a recent compilation of quantitative genetics estimates obtained from wild populations by Postma (2014) showed that for estimates obtained with animal models, the median number of records for life history traits was 377 (range 6–4992; from 39 studies, covering 19 species) and 363 for morphological traits (range 50–38 024; from 47 studies, covering 22 species). This suggests that sample size alone is not sufficient to infer pedigree quality, as it does not reflect the connectivity of pedigrees (Wilson et al., 2010). In our study system, the low recruitment of nestlings (1.3%) results in very few grandparent links within our pedigree, which greatly reduces its power. However, this problem is far less important for nestling traits as the high-EPP rate results in a genetic pedigree containing several half-sib families, which increase its power (see GG versus SS, Figure 2c) despite a smaller number of observations compared with the social pedigree for all categories (Table 1). Thus, given that pedigree structures are different from one study system to another, their impact on reliability of quantitative genetic estimates is best predicted from simulation analyses.
Various ecological and population specific factors could generate fluctuations in the precision and accuracy of quantitative genetic estimates among study systems. Here, our study system was characterized by a high rate of EPP and a low recruitment, two characteristics that affected the reliability of quantitative genetic estimates for some trait types. Although these characteristics are certainly common in other migratory species (for example, Common reed bunting (Emberiza schoeniclus) and Red-eyed vireo (Vireo olivaceus), Griffith et al., 2002), other characteristics, such as low-recapture rate between years in open study systems, a sex bias in sampling or large natal dispersal may also have important consequences on the precision of estimates. Moreover, differential mortality or capture rate might bias the sampling of some traits, and consequently affect the accuracy of estimates that can be obtained (for example, Dingemanse et al., 2009). It is thus important to appropriately identify the limits in a particular system and assess their impacts on the reliability of quantitative genetic estimates by using simulations.
Caution needed with model parameterization
The choice of which variance terms to include in a model is a crucial step that can have substantial impacts on reliability of genetic variance estimates (Kruuk and Hadfield, 2007; Wilson, 2008). Here, the decision to include or not a particular variance term relied on LRTs, which is a widely used approach (Wilson et al., 2010). However, in some cases even if the inclusion of a particular component is not improving the model likelihood, it may still have to be included (Wilson et al., 2010). This is the case for component of variance attributable to repeated measurement of a given individual (VPE). In our models, we had to account for multiple observations by fitting a VPE term in our model, but this component was almost completely confounded with VA. This situation probably occurred because of the low number of observations per individual (mean of 1.5 (averaged across traits; range: 1–7) for morphological traits and 1.2 (range: 1–7) for reproductive traits), a situation that should be present in other short-lived species datasets. Not accounting for repeated measurements in an animal model is equivalent to considering each repeated measurement as a clone of the same individual, which is inappropriate. In such cases, using a dataset with only one observation per individual may lead to more accurate estimates and easier model convergence.
Standard errors are often viewed as predictors of an estimate’s accuracy or significance, but this can be misleading (Krzywinski and Altman, 2013). Estimates from animal models have generally smaller standard errors than those obtained with parent–offspring regressions (Kruuk, 2004; Postma, 2014), partly due to their integration of multiple observations for a given individual (Åkesson et al., 2008). As previously stated, failing to account for multiple observations can create unpredictable bias in phenotypic variance component estimates (Kruuk and Hadfield, 2007) and considerably reduce standard errors around these biased estimates (Supplementary Figure D6). Moreover, it seems that problems of identifiability could also result in reduced standard errors. For example, in our empirical analyses, small standard errors were estimated around h2 estimates for reproductive traits from univariate animal models with repeated measurements per individual (SE range: 0.03–0.04), which could have led us to misleadingly conclude that our dataset was powerful and our estimates were precise for these traits.
This study was performed with a frequentist method (that is, REML) rather than a Bayesian method (that is, MCMC) because it generally gives more precise and accurate estimates for traits following a Gaussian distribution and it necessitates a shorter computation time (de Villemereuil et al., 2013). Bayesian methods have other advantages, such as the use of posterior distributions for the calculation of derived parameters (for example, heritability) and associated standard errors and they are particularly useful for analyses of non-Gaussian traits (Morrissey et al., 2014). In the present context, problems related to missing of an important source of resemblance among individuals are not method specific and could impact any model regardless of the method used for its estimation. However, it is less clear if identifiability problems could be better uncovered with Bayesian approach, for example through an appropriate MCMC checking. Simulation studies such as this one could be applied to animal models resolved with a Bayesian approach to better assess its limits.
Impact of high level of EPP on estimates
In theory, EPPs, if not accounted for, could downwardly bias additive genetic variance and resulting heritability estimates (Charmantier and Réale, 2005). Yet, previous simulation studies showed that even if biases increased with the importance of EPPs, with increasing heritability and when focal traits were directly related to the number of extra-pair young produced, underestimations were generally smaller than 15% (Firth et al., 2015). In our study, the rate of EPP was higher than those previously tested so far (up to 40% in Charmantier and Réale, 2005, 12.5% in Firth et al., 2015), but its effect on quantitative genetics estimates was complex. First, our simulation analyses showed that the bias in h2 estimates resulting from using the social instead of the genetic pedigree changed depending on the trait category (that is, higher for nestling traits). Also, our empirical analyses suggested that the impact of not accounting for EPP was noticeable for morphological and nestling traits, but sometimes resulted in higher h2 and CVA when using the social pedigree (that is, for nestling wing length and body mass). A similar unexpected finding was previously reported in blue tits (Cyanistes caeruleus), where h2 estimates from a social pedigree were sometimes higher than those from a genetic pedigree (Charmantier and Réale, 2005). These positive biases observed in empirical data could be due to the influence of the social father (for example through parental care), which could be captured in VA components (Griffith et al., 1999; Charmantier and Réale, 2005) rather than due to pedigree errors. To assess this possible problem, we performed an additional animal model analysis including mother and social father identities as additional variance components. We found that new h2 and CVA values obtained using the social pedigree were smaller than values obtained with the genetic one (change in estimates for body mass h2=−0.06, CVA=−0.011; wing length h2=−0.04, CVA=−0.041; difference calculated with univariate animal models; see Supplementary Table C5 for details). This further emphasizes the importance of model parameterization on reliability of variance component estimates. Finally, even if the genetic pedigree better represents relationships among individuals than the social pedigree, it probably still contains a few incorrect links among individuals and thus the estimated heritabilities might also be slightly downwardly biased.
Conclusion
Simulation analyses are now widely accessible to anyone with minimal programming skills in R, for instance by using the ‘phensim’ function within the package PEDANTICS, the ‘rbv’ function within the package MCMCglmm (Hadfield, 2010) or the ‘drfx’ and the ‘grfx’ function within the package NADIV (Wolak, 2012). Moreover, other methods such as profile likelihoods (Meyer, 2008; implemented in the ‘proLik’ function within the package NADIV, Wolak, 2012) are more accessible and can also improve our comprehension of a particular dataset limits (Houle and Meyer, 2015). As each study system is unique, in terms of pedigree structure, number of repeated measures per individual or potential causes of pedigree errors, it is difficult to predict the reliability of quantitative genetics estimates without testing it formally. Detailed simulations should be routinely included when reporting quantitative genetics analyses performed within a new study system, or when applying more complex modeling to previously studied populations, to explicitly assess the precision and accuracy of genetic variance components (Morrissey et al., 2007).
Data archiving
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.n8c57.
References
Åkesson M, Bensch S, Hasselquist D, Tarka M, Hansson B, Akesson M . (2008). Estimating heritabilities and genetic correlations: comparing the ‘animal model’ with parent–offspring regression using data from a natural population. PLoS One 3: e1739.
Bérénos C, Ellis PA, Pilkington JG, Pemberton JM . (2014). Estimating quantitative genetic parameters in wild populations: a comparison of pedigree and genomic approaches. Mol Ecol 23: 3434–3451.
Blows MW, Hoffmann AA . (2005). A reassessment of genetic limits to evoutionary change. Ecology 86: 1371–1384.
Bolker B . (2008) Ecological Models and Data in R. Princeton, University Princeton, NJ, USA.
Charmantier A, Réale D . (2005). How do misassigned paternities affect the estimation of heritability in the wild? Mol Ecol 14: 2839–2850.
Dingemanse NJ, Kazem AJN, Réale D, Wright J . (2009). Behavioural reaction norms: animal personality meets individual plasticity. Trends Ecol Evol 25: 81–89.
Dunn PO, Whittingham LA, Pitcher TE . (2001). Mating systems, sperm competition, and the evolution of sexual dimorphism in birds. Evolution 55: 161–175.
Falconer DS, Mackay TFC . (1996) Introduction to Quantitative Genetics, 4th edn. Pearson: Essex, UK.
Firth JA, Hadfield JD, Santure AW, Slate J, Sheldon BC . (2015). The influence of nonrandom extra-pair paternity on heritability estimates derived from wild pedigrees. Evolution 69: 1336–1344.
Garcia-Gonzalez F, Simmons LW, Tomkins JL, Kotiaho JS, Evans JP . (2012). Comparing evolvabilities: common errors surrounding the calculation and use of coefficients of additive genetic variation. Evolution 66: 2341–2349.
Ghilain A, Bélisle M . (2008). Breeding success of tree swallows along a gradient of agricultural intensification. Ecol Appl 18: 1140–1154.
Griffith SC, Owens IPF, Burke T . (1999). Environmental determination of a sexually selected trait. Science 400: 358–360.
Griffith SC, Owens IPF, Thuman KA . (2002). Extra pair paternity in birds: a review of interspecific variation and adaptive function. Mol Ecol 11: 2195–2212.
Hadfield JD . (2010). MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J Stat Softw 33: 1–22.
Henderson CR . (1984) Applications of Linear Models in Animal Breeding. University of Guelph: Guelph, ON, Canada.
Hendry AP, Kinnison MT, Heino M, Day T, Smith TB, Fitt G et al. (2011). Evolutionary principles and their practical application. Evol Appl 4: 159–183.
Hoffmann AA, Merilä J . (1999). Heritable variation and evolution under favourable and unfavourable conditions. Trends Ecol Evol 14: 96–101.
Hosner PA, Winkler DW . (2007). Dispersal distances of tree swallows estimated from continent-wide and limited-area data. J Field Ornithol 78: 290–297.
Houle D . (1992). Comparing evolvability and variability of quantitative traits. Genetics 130: 195–204.
Houle D, Meyer K . (2015). Estimating sampling error of evolutionary statistics based on genetic covariance matrices using maximum likelihood. J Evol Biol 28: 1542–1549.
Husby A, Kawakami T, Rönnegård L, Smeds L, Ellegren H, Qvarnström A . (2015). Genome-wide association mapping in a wild avian population identifies a link between genetic and phenotypic variation in a life-history trait. Proc R Soc B 282: 20150156.
Hussell DJT . (1983). Age and plumage color in female tree swallows. J Field Ornithol 54: 312–318.
Kalinowski ST, Taper ML, Marshall TC . (2007). Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol 16: 1099–1106.
Kruuk LEB . (2004). Estimating genetic parameters in natural populations using the ‘animal model’. Philos Trans R Soc B 359: 873–890.
Kruuk LEB, Hadfield JD . (2007). How to separate genetic and environmental causes of similarity between relatives. J Evol Biol 20: 1890–1903.
Kruuk LEB, Slate J, Wilson AJ . (2008). New answers for old questions: the evolutionary quantitative genetics of wild animal populations. Annu Rev Ecol Evol Syst 39: 525–548.
Krzywinski M, Altman N . (2013). Points of significance: error bars. Nat Methods 10: 921–922.
Lande R . (1981). The minimum number of genes contributing to quantitative variation between and within populations. Genetics 99: 541–553.
Lande R, Arnold SJ . (1983). The measurement of selection on correlated characters. Evolution 37: 1210–1226.
Lee C, Pollak EJ . (1997). Influence of sire misidentification on sire x year interaction variance and direct-maternal genetic covariance for weaning weight in beef cattle. J Anim Sci 75: 2858–2863.
Lemons PR, Marshall TC, McCloskey SE, Sethi SA, Schmutz JA, Sedinger JS . (2015). A likelihood-based approach for assessment of extra-pair paternity and conspecific brood parasitism in natural populations. Mol Ecol Resour 7: 107–116.
Lessard A, Bourret A, Bélisle M, Pelletier F, Garant D . (2014). Individual and environmental determinants of reproductive success in male tree swallow (Tachycineta bicolor. Behav Ecol Sociobiol 68: 733–742.
Lynch M, Walsh B . (1998) Genetics and Analysis of Quantitative Traits. Sinauer Associates: Sunderland, MA, USA.
Meyer K . (2008). Likelihood calculations to evaluate experimental designs to estimate genetic variances. Heredity 101: 212–221.
Morrissey MB, Kruuk LEB, Wilson AJ . (2010). The danger of applying the breeder’s equation in observational studies of natural populations. J Evol Biol 23: 2277–2288.
Morrissey MB, de Villemereuil P, Doligez B, Gimenez O . (2014). Bayesian approaches to the quantitative genetic analysis of natural populations. In: Charmantier A, Garant D et al. (eds) Quantitative Genetics in the Wild. Oxford University Press, Oxford, UK, pp 228–253.
Morrissey MB, Wilson AJ . (2010). PEDANTICS: an R package for pedigree-based genetic simulation and pedigree manipulation, characterization and viewing. Mol Ecol Resour 10: 711–719.
Morrissey MB, Wilson AJ, Pemberton JM, Ferguson MM . (2007). A framework for power and sensitivity analyses for quantitative genetic studies of natural populations, and case studies in Soay sheep (Ovis aries. J Evol Biol 20: 2309–2321.
Postma E . (2014). Four decades of estimating heritabilities in wild vertebrate populations: improved methods, more data, better estimates? In: Charmantier A, Garant D et al. (eds) Quantitative Genetics in the Wild. Oxford University Press, Oxford, UK, pp 16–33.
Quinn JL, Charmantier A, Garant D, Sheldon BC . (2006). Data depth, data completeness, and their influence on quantitative genetic estimation in two contrasting bird populations. J Evol Biol 19: 994–1002.
Core Team R . (2015). R: a language and environment for statistical computing http://www.R-project.org/.
Via S, Lande R . (1985). Genotype-environment interaction and the evolution of phenotypic plasticity. Evolution 39: 505–522.
de Villemereuil P, Gimenez O, Doligez B . (2013). Comparing parent–offspring regression with frequentist and Bayesian animal models to estimate heritability in wild populations: A simulation study for Gaussian and binary traits. Methods Ecol Evol 4: 260–275.
Vinkhuyzen AAE, Wray NR, Yang J, Goddard ME, Visscher PM . (2013). Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu Rev Genet 47: 75–95.
Walther BA, Moore JL . (2005). The concepts of bias, precision and accuracy, and their use in testing the performance of species richness estimators, with a literature review of estimator performance. Ecography 28: 815–829.
Wilson AJ . (2008). Why h2 does not always equal VA/VP? J Evol Biol 21: 647–650.
Wilson AJ, Morrissey MB, Adams MJ, Walling CA, Guinness FE, Pemberton JM et al. (2011). Indirect genetics effects and evolutionary constraint: an analysis of social dominance in red deer, Cervus elaphus. J Evol Biol 24: 772–783.
Wilson AJ, Réale D, Clements MN, Morrissey MM, Postma E, Walling CA et al. (2010). An ecologist’s guide to the animal model. J Anim Ecol 79: 13–26.
Winkler D, Hallinger K, Ardia D, Robertson R, Stutchbury B, Cohen R . (2011). Tree swallow (Tachycineta bicolor. Birds North Am Online. http://bna.birds.cornelledu/bna/. Accessed on 1 Oct 2013.
Wolak ME . (2012). nadiv : an R package to create relatedness matrices for estimating non-additive genetic variances in animal models. Methods Ecol Evol 3: 792–796.
Wolak ME, Roff DA, Fairbairn DJ . (2015). Are we underestimating the genetic variances of dimorphic traits? Ecol Evol 5: 590–597.
Acknowledgements
We thank the 40 farm owners for providing access to their lands and all graduate students and field and laboratory assistants who have contributed in gathering data in our system over the years. We also thank Fanie Pelletier and Marc Bélisle for their long-term involvement in the southern Québec Tree swallow study. We also thank them as well as Denis Réale, Matthew Wolak, Michael Morrissey and two anonymous reviewers for constructive comments on a previous version of this manuscript. Animals were captured and handled in compliance with the Canadian Council on Animal Care, under the approval of the Université de Sherbrooke Animal Ethics Committee (Protocol Number: DG2010-01 and DG2014-01-Université de Sherbrooke). This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) discovery grants to DG, Fanie Pelletier and Marc Bélisle and by the Canada Research Chairs program to Fanie Pelletier and Marc Bélisle. AB was supported by a postgraduate NSERC scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Bourret, A., Garant, D. An assessment of the reliability of quantitative genetics estimates in study systems with high rate of extra-pair reproduction and low recruitment. Heredity 118, 229–238 (2017). https://doi.org/10.1038/hdy.2016.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.92
This article is cited by
-
The effect of repeated measurements and within-individual variance on the estimation of heritability: a simulation study
Behavioral Ecology and Sociobiology (2024)
-
Heritability of plumage colour morph variation in a wild population of promiscuous, long-lived Australian magpies
Heredity (2019)
