
Abstract
It has been well documented that variants in genes encoding centrosomal proteins cause primary autosomal recessive microcephaly, although the association between centrosomal defects and the etiology of microcephaly syndromes is not fully understood. Polo-like kinase 4 (PLK4) is one of the centrosomal proteins required for centriole duplication. We here describe a patient with microcephaly and chorioretinopathy that harbors compound heterozygous missense variants, c.[442A>G]; [2336G>A], in the PLK4 gene. One of these variants, c.442A>G (p.(M148V)), resides in the kinase domain, and the other, c.2336G>A (p.(C779Y)), in the polo-box domain. Aberrant spindle formation was observed in a LCL derived from this patient. Overexpression experiments of the variant PLK4 proteins demonstrated that the p.(C779Y) but not the p.(M148V) had lost centriole overduplication ability. The altered mobility pattern of both variant proteins on a western blot further suggested alterations in post-translation modification. Our data lend support to the hypothesis that impaired centriole duplication caused by PLK4 variants may be involved in the etiology of microcephaly disorder.
Similar content being viewed by others

Introduction
Autosomal recessive microcephaly and chorioretinopathy (MCCRP) is a developmental disorder characterized by primary microcephaly, delayed psychomotor development, growth retardation with dwarfism and visual impairment. Recently, variants in the PLK4 gene, one of the key regulators of centriole duplication, were identified in patients with microcephaly, growth failure and retinopathy1, 2 (MCCRP2 [MIM616171]).
In our current study, we describe a female MCCRP patient with missense variants in PLK4. She was affected by autosomal recessive microcephaly and developmental eye disease that were consistent with the MCCRP phenotype. Cell biological analyses using a lymphoblastoid cell line (LCL) derived from this patient and functional analyses of the variant PLK4 proteins were performed to examine the impacts on centriole biosynthesis.
Materials and methods
Patient
The genetic testing used in this study was approved by the ethical committees of Fujita Health University in accordance with the principles of the Declaration of Helsinki, and the Ethical Guidelines for Human Genome/Gene Analysis Research by the Ministry of Education, Culture, Science, and Technology, the Ministry of Health, Labor, and Welfare, and the Ministry of Economy, Trade, and Industry of Japan. Blood samples from the affected individual and her parents were obtained with informed consent in accordance with local institutional review board guidelines.
Whole-exome sequencing and validation
Genomic DNA extraction from the peripheral blood of the patient and parents and whole-exome sequencing and validation were performed as previously described.3 The identified PLK4 variants were submitted to the LOVD database at http://www.LOVD.nl/PLK4 (individual ID: 00072170).
Cell culture
An Epstein–Barr virus transformed LCL line was established from peripheral blood mononuclear cells and cultured in RPMI 1640 containing 10% FBS established from a healthy control also. Asynchronous growing cells were fixed with ice-cold methanol to analyze the number of centrosomes and mitotic spindle formation by immunofluorescence.
Transfection experiments
The cDNA encoding wild-type (WT) human PLK4 was chemically synthesized and cloned into the pEGFP-C1 vector. Mutations were generated by PCR using the WT construct as a template. All constructs were verified by DNA sequencing and transfected into HeLa cells. At 24 h posttransfection, cell lysates were prepared for western blot analysis, or the cells were fixed with ice-cold methanol for immunofluorescence analysis.
Western blotting
Whole-cell lysate of the asynchronously growing LCL and recombinant proteins expressed in HeLa cells were analyzed by western blot followed by image analysis performed as described previously.4 Rabbit anti-PLK4 (1:500; Proteintech, Rosemont, IL, USA), mouse anti-ACTB (AC-15) (1:500000; Sigma, St Louis, MO, USA) or rabbit anti-GFP (1:500; MBL, Nagoya, Japan) was used as the primary antibody. HRP-conjugated anti-rabbit IgG or anti-mouse IgG (Thermo Scientific, Rockford, IL) was used as the secondary antibody.
Immunofluorescence
The fixed LCL or HeLa cells were stained with primary antibodies at the following dilutions: rabbit anti-GFP (1:500), mouse anti-α-tubulin (1:250; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-γ-tubulin (1:1000; Sigma), rabbit polyclonal anti-phospho Histone H3 (pH3) (Ser10) antibody (1:100; Merck Millipore, Darmstadt, Germany). The secondary antibodies used were donkey anti-rabbit IgG Alexa Fluor 488, donkey anti-rabbit IgG Alexa Fluor 594, donkey anti-mouse IgG Alexa Fluor 488 and donkey anti-mouse IgG Alexa Fluor 594 (Life Technologies, Carlsbad, CA, USA) at a 1:1000 dilution. Image acquisition was performed as previously described.3 For centrosome scoring, we counted the γ-tubulin signals of the LCL and HeLa mitotic cells. Spindle formation of the mitotic LCL was classified into four types in the similar way of the previous study.1 Bipolar spindles were normal, monopolar cells had only one spindle pole, multipolar cells had excess spindle poles and disorganized cells failed to establish a spindle pole.
Results
Patient characteristics
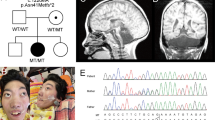
The study patient was a female born at a gestational age of 37 weeks. Her parents were healthy and non-consanguineous. Her birth weight was 1322 g (−3.9 SD), head circumference was 20.6 cm (−7.3 SD), and body length was 38.0 cm (−5.1 SD). She had microcephaly, bilateral microphthalmos and persistent hyperplastic primary vitreous of her left eye. Brain MRI revealed trigonocephaly, microcephaly with simplified gyri, colpocephaly and bilateral asymmetric periventricular nodular heterotopia (Figure 1A). Her karyotype was normal (46,XX).

(A) Brain MRI of the patient at the age of 6 months. a and b: T1-weighted images. c and d: T2-weighted images. (B) Filtering steps for determining candidate variants in the patient. The top row is the number of called variants by next-generation sequencing. The second row shows the number of variants remaining after filtering out known variants in the databases, except for those that are also known as variants affecting function. The third row indicates the number of variants remaining after excluding synonymous change variants. The bottom row indicates the number of variants consistent with the phenotype in the pedigree (ie, the total number of autosomal recessive, X-linked recessive, de novo and compound heterozygous variants). (C) Validation of the PLK4 variants by Sanger sequencing.
Her developmental quotient at 2 years of age, measured using the Tsumori-Inage Developmental Questionnaire, was 21. Her development was considered to be severely delayed. At the age of 32 months, her weight was 6440 g (−4.2 SD), head circumference was 34.7 cm (−8.9 SD), and body length was 67.6 cm (−6.8 SD).
Identification of variants in PLK4
We performed whole-exome sequencing analysis of this patient and identified novel compound heterozygous variants, c.[442A>G]; [2336G>A], in the PLK4 gene (NG_041821.1; NM_014264.4; Figure 1B and C and Supplementary Table S1). Subsequent Sanger sequencing confirmed these variants (Figure 1C). The c.442A>G (p.(M148V)) variant was inherited from the healthy mother, and the c.2336G>A (p.(C779Y)) variant from the healthy father. Both variants were predicted to be damaging by Polyphen-2 and SIFT (Supplementary Table S1). No variants related to microcephaly were identified in any other genes in the patient (Supplementary Table S1). M148 and C779 residues are highly conserved amino acids in the PLK4 protein across species. The M148 residue is located in the kinase domain, whereas the C779 residue is located in the second of the three polo-box domains, PB2. The C779 residue is defined as a homodimer interface by the NCBI Conserved Domain Database (accession number cd13115; http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml5).
Abnormal centrosome number and impaired mitotic spindle formation in the study patient
PLK4 protein level of an LCL derived from the patient was found to be comparable to that of control by western blot (Figure 2a). To examine whether centriole biosynthesis was impaired in our patient, the LCL was used to analyze the centrosome number. Asynchronously growing cells were fixed and stained with anti-γ-tubulin antibodies (Figure 2b). In mitotic cells, the normal centrosome number is theoretically 2. However, 16.7% of the cells from our patient had 1 or ≥3 centrosomes (Figure 2c). This rate was slightly higher than that of the control cells (14.7%, Figure 2c).

PLK4 expression, centrosome number and spindle formation in LCL. (a) Western blot analysis of PLK4 protein in LCL derived from a control and the patient. (b) Representative immunofluorescent microscopic images of the mitotic LCL derived from the patient. γ-tubulin (green) is a centrosome marker. Phospho-histone H3 (red) was used as a mitotic marker. The centrosome number is indicated in each image. Scale bar, 10 μm. (c) Proportion of cells with the indicated centrosome number. (d) Representative immunofluorescent microscopic images of the mitotic LCL derived from the patient. α-tubulin (green) is a spindle marker. Spindle morphology is indicated in each image. (e) Proportion of cells with the indicated spindle morphology (*P<0.05, two-tailed Student’s t-test). The results in c and e were obtained from three independent experiments shown in b and d, respectively. n=50 cells in each experiment. Data are the average value±SEM.
We also analyzed spindle polarity of the mitotic cells because the centrosome plays a role in the formation of bipolar mitotic spindles. α-tubulin labeling of the LCL from our patient revealed that 25% of the mitotic cells had an aberrant spindle morphology, whereas >90% of the control mitotic cells were bipolar (Figure 2d and e). Of note, cells with a monopolar spindle were increased in our patient. Other morphological defects, such as excess spindle poles or a disorganized spindle, were more frequent in our patient than in the control cells (Figure 2e). These results suggest that spindle formation was impaired in our patient because of a centriole duplication abnormality.
Centriole amplification abilities of the variant PLK4 proteins
We next examined the properties of PLK4 proteins harboring the p.(M148V) and p.(C779Y) variants. It is known that the overexpression of PLK4 induces centriole amplification.6, 7 EGFP-tagged variant proteins were overexpressed in HeLa cells and analyzed by immunofluorescence (Figure 3). When WT PLK4 was overexpressed in HeLa cells, centriole amplification was observed and >70% of the cells had excess centrosomes (Figure 3a and b). In contrast, overexpression of the p.(C779Y) variant had no effect on centriole amplification, suggesting a loss of function in centriole duplication. On the other hand, overexpression of the p.(M148V) proteins could amplify the centrioles and produce excess centrosomes to the same extent as WT PLK4 (Figure 3a and b). Intense immunofluorescent signals corresponding to both EGFP-tagged WT and the p.(M148V) variant PLK4 were detected at the centrioles in the centrosomes, whereas the p.(C779Y) PLK4 signals were weak in the centrosomes and found to be predominantly diffused within the cytoplasm (Figure 3a). These results suggest that the p.(C779Y) variant within the PB2 domain of PLK4 prevented its localization to the centrioles leading to a dysfunction in centriole duplication.

Overexpression analysis of the PLK4 variants. (a) Representative immunofluorescent microscopic images of HeLa cells transfected with the indicated EGFP-tagged PLK4 expression plasmids. γ-tubulin (centrosome) and EGFP (centriole) are shown in red and green, respectively. Centrosomes in the boxes are magnified. Scale bar, 10 μm. (b) Proportion of cells with more than two centrosomes were examined from the experiments shown in (a). The results of three independent experiments are shown. n=50 cells in each experiment. Data are average values±SEM (**P<0.01, two-tailed Student's t-test). (c) Western blot analysis using an anti-GFP antibody. Asterisk indicates the normal band size estimated from a molecular weight calculation of EGFP-PLK4. Values in parentheses indicate the intensity ratio of the larger band to the normal band.
Western blot analysis of HeLa lysates transfected with the EGFP-tagged PLK4 expression plasmids was performed using an anti-GFP antibody (Figure 3c). An ~136 kDa band was detected for each construct consistent with the calculated molecular weight of the EGFP-tagged PLK4. In addition to the 136 kDa band, higher molecular weight bands of ~145 kDa were also detectable with WT and the p.(M148V) variant, although the intensity of these bands was remarkably reduced for the p.(C779Y) variant. These higher molecular weight bands are likely to originate from the post-translation modification of the PLK4 protein. The ratio of the band intensity of the ~145 kDa to the 136 kDa p.(M148V) products was twofold lower than that of the WT proteins (Figure 3c). Although the origin of this band shift has not been identified, this result indicates that the p.(M148V) variant is not functionally equivalent to WT PLK4. Because the intensity of the higher molecular weight band is considerably weaker for the p.(C779Y) PLK4, which lacks centriole duplication ability, we speculate that the p.(M148V) variant may also have an effect on the centriole function of PLK4, and these two variants may thus coordinately produce the microcephaly phenotype of our study patient.
Discussion
In our present study, we have identified novel compound heterozygous missense PLK4 variants in a patient with microcephaly, microphthalmos and persistent hyperplastic primary vitreous. Autosomal recessive MCCRP is characterized by delayed psychomotor development and visual impairment, often accompanied by short stature. To date, three responsible genes have been described. MCCRP1 is caused by homozygous or compound heterozygous variants in the TUBGCP6 gene,1 MCCRP2 [MIM616171] is caused by homozygous truncating variant in the PLK4 gene,1 and MCCRP3 is caused by compound heterozygous variants in the TUBGCP4 gene.8 The features of the visual impairment in MCCRP patients are variable, but are typically characterized by punched-out retinal lesions and occasional retinal folds leading to retinal detachment.8 A persistent hyperplastic primary vitreous is included in the chorioretinopathy caused by the mutations in KIF11.9 The clinical features of our current patient accorded with those of MCCRP.
PLK4 is one of the key regulators of centriole duplication.6, 10 PLK4 variants thus possibly produce the MCCRP phenotype in a similar way to MCPH and Seckel syndrome caused by variants in genes encoding centrosomal proteins.11 The prevailing hypothesis is that the variants in the centrosome genes impair centriole duplication and reduce mitosis efficiency, thereby inducing cell death or reduced cellular proliferation during early embryogenesis and leading to dwarfism. In support of this possibility, neurogenic cells with impaired regulation of centriole duplication are predisposed to mitotic delay or chromosomal aneuploidy leading to a microcephaly and/or retinal phenotype in the patient.12 Indeed, a mouse model of centrosome amplification manifests microcephaly.13
PLK4 has a kinase domain at its amino terminal and three PB domains mediating centriole localization and homodimerization at its carboxyl terminal.14 PLK4 autophosphorylation regulates its activity,15, 16 which is strictly controlled in terms of timing and location during the cell cycle.17 We have shown from our current analyses that the p.(C779Y) PLK4 variant lacks the normal function in centriole duplication. It is thus probable that the p.(C779Y) variant in the PB2 domain impedes the homodimerization of PLK4 and consequently the loss of normal protein function due to the prevalence of the monomeric form.16 On the other hand, the p.(M148V) variant in the kinase domain of PLK4 could amplify the number of centrioles at WT levels when overexpressed in HeLa cells. It is therefore possible that the overexpressed p.(M148V) variant does not reduce the overall kinase activity levels of PLK4 in the presence of the WT protein even if it lacks kinase activity itself.18 Western blot analysis of overexpressed PLK4 suggests that the p.(M148V) variant has slightly less activity than WT. Although the exact function of the M148 residue has not yet been characterized, the strong evolutionary conservation of this residue suggests that it has functional importance.
Finally, previous studies of Plk4 mutant mice have revealed that a homozygous knockout or mutation in the kinase domain is embryonic lethal whilst a haploinsufficiency of PLK4 appears to produce no phenotype other than cancer susceptibility.19, 20, 21 Hence, our current study patient likely survived due to the residual marginal activity of PLK4. We speculate that the reduced activity of the p.(M148V) variant rather than a loss-of-function prevented embryonic lethality. The variants we have here described provide a better understanding of the clinical and biological relevance of PLK4 to centriole duplication.
References
Martin CA, Ahmad I, Klingseisen A et al: Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet 2014; 46: 1283–1292.
Shaheen R, Al Tala S, Almoisheer A, Alkuraya FS : Mutation in PLK4, encoding a master regulator of centriole formation, defines a novel locus for primordial dwarfism. J Med Genet 2014; 51: 814–816.
Yokoi S, Ishihara N, Miya F et al: TUBA1A mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci Rep 2015; 5: 15165.
Tsutsumi M, Kogo H, Kowa-Sugiyama H et al: Characterization of a novel mouse gene encoding an SYCP3-like protein that relocalizes from the XY body to the nucleolus during prophase of male meiosis I. Biol Reprod 2011; 85: 165–171.
Marchler-Bauer A, Derbyshire MK, Gonzales NR et al: CDD: NCBI's conserved domain database. Nucleic Acids Res 2015; 43: D222–D226.
Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA : The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 2005; 7: 1140–1146.
Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA : Plk4-induced centriole biogenesis in human cells. Dev Cell 2007; 13: 190–202.
Scheidecker S, Etard C, Haren L et al: Mutations in TUBGCP4 alter microtubule organization via the γ-tubulin ring complex in autosomal-recessive microcephaly with chorioretinopathy. Am J Hum Genet 2015; 96: 666–674.
Jones GE, Ostergaard P, Moore AT et al: Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet 2014; 22: 881–887.
Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L et al: SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 2005; 15: 2199–2207.
Verloes A, Drunat S, Gressens P, Passemard S : Primary autosomal recessive microcephalies and seckel syndrome spectrum disorders. In: Pagon RA et al (eds): Gene Reviews. Seatle, WA, USA: University of Washington, 1993.
Chavali PL, Pütz M, Gergely F : Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci 2014; 369: pii: 20130468.
Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R : Centrosome amplification causes microcephaly. Nat Cell Biol 2013; 15: 731–740.
Slevin LK, Nye J, Pinkerton DC, Buster DW, Rogers GC, Slep KC : The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure 2012; 20: 1905–1917.
Cunha-Ferreira I, Bento I, Pimenta-Marques A et al: Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr Biol 2013; 23: 2245–2254.
Klebba JE, Buster DW, McLamarrah TA, Rusan NM, Rogers GC : Autoinhibition and relief mechanism for Polo-like kinase 4. Proc Natl Acad Sci USA 2015; 112: E657–E666.
Sillibourne JE, Bornens M : Polo-like kinase 4: the odd one out of the family. Cell Div 2010; 5: 25.
Guderian G, Westendorf J, Uldschmid A, Nigg EA : Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J Cell Sci 2010; 123: 2163–2169.
Hudson JW, Kozarova A, Cheung P et al: Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr Biol 2001; 11: 441–446.
Ko MA, Rosario CO, Hudson JW et al: Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat Genet 2005; 37: 883–888.
Harris RM, Weiss J, Jameson JL : Male hypogonadism and germ cell loss caused by a mutation in Polo-like kinase 4. Endocrinology 2011; 152: 3975–8395.
Acknowledgements
We thank E. Hosoba for technical assistance. This study was supported in part by a grant for Research on Applying Health Technology from the Ministry of Health, Labour and Welfare of Japan to FM, NO, MK, MY, YK, KK and SS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Tsutsumi, M., Yokoi, S., Miya, F. et al. Novel compound heterozygous variants in PLK4 identified in a patient with autosomal recessive microcephaly and chorioretinopathy. Eur J Hum Genet 24, 1702–1706 (2016). https://doi.org/10.1038/ejhg.2016.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.119
This article is cited by
-
Genetic interaction between PLK1 and downstream MCPH proteins in the control of centrosome asymmetry and cell fate during neural progenitor division
Cell Death & Differentiation (2022)
-
Transmission ratio distortion of mutations in the master regulator of centriole biogenesis PLK4
Human Genetics (2022)
-
Overexpression of the PLK4 Gene as a Novel Strategy for the Treatment of Autosomal Recessive Microcephaly by Improving Centrosomal Dysfunction
Journal of Molecular Neuroscience (2021)
-
A female patient with retinoblastoma and severe intellectual disability carrying an X;13 balanced translocation without rearrangement in the RB1 gene: a case report
BMC Medical Genomics (2019)
