
Abstract
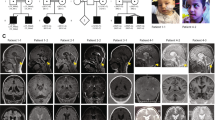
Pontocerebellar hypoplasia (PCH) is currently classified into 16 subgroups. Using mostly next-generation sequencing, pathogenic variants have been identified in as many as 24 PCH-associated genes. PCH type 8 (PCH8) is a rare heterogeneous disorder. Its clinical presentation includes severe development delay, increased muscle tone, microcephaly, and magnetic resonance imaging (MRI) abnormalities such as reduced cerebral white matter, a thin corpus callosum, and brainstem and cerebellar hypoplasia. To date, only two variants in the CHMP1A gene (MIM: 164010), NM_002768.5: c.88 C > T (p.Glu30*) and c.28–13 G > A, have been identified homozygously in seven patients with PCH8 from four families (MIM: 614961). CHMP1A is a subunit of the endosomal sorting complex required for transport III (ESCRT-III), which regulates the formation and release of extracellular vesicles. Biallelic CHMP1A loss of function impairs the ESCRT-III-mediated release of extracellular vesicles, which causes impaired progenitor proliferation in the developing brain. Herein, we report a patient with PCH8 who had a homozygous CHMP1A variant, c.122delA (p.Asn41Metfs*2), which arose from segmental uniparental disomy. Although our patient had similar MRI findings to those of previously reported patients, with no progression, we report some novel neurological and developmental findings that expand our knowledge of the clinical consequences associated with CHMP1A variants.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


Change history
24 January 2023
A Correction to this paper has been published: https://doi.org/10.1038/s10038-023-01125-5
References
Nuovo S, Micalizzi A, Romaniello R, Arrigoni F, Ginevrino M, Casella A, et al. Refining the mutational spectrum and gene-phenotype correlates in pontocerebellar hypoplasia: results of a multicentric study. J Med Genet. 2022;59:399–409.
Appelhof B, Wagner M, Hoefele J, Heinze A, Roser T, Koch-Hogrebe M, et al. Pontocerebellar hypoplasia due to bi-allelic variants in MINPP1. Eur J Hum Genet. 2021;29:411–21.
van Dijk T, Baas F, Barth PG, Poll-The BT. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis. 2018;13:92.
Juan T, Furthauer M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin Cell Dev Biol. 2018;74:66–77.
McCullough J, Colf LA, Sundquist WI. Membrane fission reactions of the mammalian ESCRT pathway. Annu Rev Biochem. 2013;82:663–92.
Coulter ME, Dorobantu CM, Lodewijk GA, Delalande F, Cianferani S, Ganesh VS, et al. The ESCRT-III protein CHMP1A Mediates Secretion Of Sonic Hedgehog On A Distinctive Subtype Of Extracellular Vesicles. Cell Rep. 2018;24:973–86.e8
Mochida GH, Ganesh VS, de Michelena MI, Dias H, Atabay KD, Kathrein KL, et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat Genet. 2012;44:1260–4.
Kahrizi K, Hu H, Hosseini M, Kalscheuer VM, Fattahi Z, Beheshtian M, et al. Effect of inbreeding on intellectual disability revisited by trio sequencing. Clin Genet. 2019;95:151–59.
Seyama R, Tsuchida N, Okada Y, Sakata S, Hamada K, Azuma Y, et al. Two families with TET3-related disorder showing neurodevelopmental delay with craniofacial dysmorphisms. J Hum Genet. 2022;67:157–64.
Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet. 2012;91:597–607.
Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184.
Uchiyama Y, Yamaguchi D, Iwama K, Miyatake S, Hamanaka K, Tsuchida N, et al. Efficient detection of copy-number variations using exome data: Batch- and sex-based analyses. Hum Mutat. 2021;42:50–65.
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593–9.
Saida K, Tamaoki J, Sasaki M, Haniffa M, Koshimizu E, Sengoku T, et al. Pathogenic variants in the survival of motor neurons complex gene GEMIN5 cause cerebellar atrophy. Clin Genet. 2021;100:722–30.
Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997;277:1109–13.
Scuffins J, Keller-Ramey J, Dyer L, Douglas G, Torene R, Gainullin V, et al. Uniparental disomy in a population of 32,067 clinical exome trios. Genet Med. 2021;23:1101–07.
Acknowledgements
This work was supported by AMED under grant numbers JP22ek0109486, JP22ek0109549, and JP22ek0109493 (NaM), JP20K07907 (SM), JP20K08164 (TM), JP21K07869 (EK), JP20K17936 (AF), JP20K16932 (KoH), JP21K15097 (YU), and JP20K17428 (NT); and the Takeda Science Foundation (TM and NaM). We thank Bronwen Gardner, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sakamoto, M., Shiiki, T., Matsui, S. et al. A novel homozygous CHMP1A variant arising from segmental uniparental disomy causes pontocerebellar hypoplasia type 8. J Hum Genet 68, 247–253 (2023). https://doi.org/10.1038/s10038-022-01098-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-022-01098-x
This article is cited by
-
A heterozygous germline deletion within USP8 causes severe neurodevelopmental delay with multiorgan abnormalities
Journal of Human Genetics (2024)
