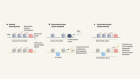
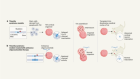
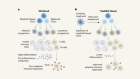
- NEWS FEATURE
Protein-slaying drugs could be the next blockbuster therapies

Illustration by David Parkins
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 567, 298-300 (2019)
doi: https://doi.org/10.1038/d41586-019-00879-3
References
Krönke, J. et al. Science 343, 301–305 (2014).
Lu, G. et al. Science 343, 305–309 (2014).
Feldman, R. M. R., Correll, C. C., Kaplan, K. B. & Deshaies, R. J. Cell 91, 221–230 (1997).
Sakamoto, K. M. et al. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Buckley, D. L. et al. Angew. Chem. Int. Ed. 51, 11463–11467 (2012).
Ito, T. et al. Science 327, 1345–1350 (2010).
Bondeson, D. P. et al. Nature Chem. Biol. 11, 611–617 (2015).
Winter, G. E. et al. Science 348, 1376–1381 (2015).
Zengerle, M., Chan, K.-H. & Ciulli, A. ACS Chem. Biol. 10, 1770–1777 (2015).
Buckley, D. L. et al. ACS Chem. Biol. 10, 1831–1837 (2015).
Lu, J. et al. Chem. Biol. 22, 755–763 (2015).
Nabet, B. et al. Nature Chem. Biol. 14, 431–441 (2018).
Deshaies, R. J. Nature Chem. Biol. 11, 634–635 (2015).
Chessum, N. E. A. J. Med. Chem. 61, 918–933 (2018).
 A father’s fight to help his sons — and fix clinical trials
A father’s fight to help his sons — and fix clinical trials
 ‘Why didn’t we think to do this earlier?’ Chemists thrilled by speedy atomic structures
‘Why didn’t we think to do this earlier?’ Chemists thrilled by speedy atomic structures
 Cancer-killing viruses show promise — and draw billion-dollar investment
Cancer-killing viruses show promise — and draw billion-dollar investment
 Open ambition
Open ambition
 The billion-dollar biotech
The billion-dollar biotech