
Abstract
The state of a diploid population segregating for two alleles at each of n loci is described by 22n genotype frequencies, or equivalently, by allele frequencies and by multilocus moments or cumulants of various orders. These measures of linkage disequilibrium cannot usually be determined, both because one cannot tell whether a gene came from the maternal or paternal gamete, and because such a large number of parameters cannot be estimated even from large samples. Simplifying assumptions must therefore be made. This paper sets out methods for estimating multilocus genotype frequencies which are appropriate for unlinked neutral loci, and for populations that are ultimately derived by mixing of two source populations. In such a hybrid population, all multilocus associations depend primarily on the number of loci involved that derive from the maternal genome, and the number derived from the paternal genome. Allele frequencies may differ across loci, and the contribution of each locus to multilocus associations may be scaled by the difference in allele frequency between source populations for that locus (δp ≤ 1). For example, the cumulant describing the association between genes i, j, k from the maternal genome, and genes i, l from the paternal genome is κi,j,k,i*l*, = δpi2 δpj δpk δpl κ3,2. The state of the population is described by n allele frequencies; n divergences, δp; and by a symmetric matrix of cumulants, κJ,K (J=0 ,..., n, K=0 ,..., n). Expressions for these cumulants under short- and long-range migration are given. Two methods for estimating the cumulants are described: a simple method based on multivariate moments, and a maximum likelihood procedure, which uses the Metropolis algorithm. Both methods perform well when tested against simulations with two or four loci.
Similar content being viewed by others

Introduction
Random mating and recombination break down associations between alleles at different genetic loci, leading populations towards “linkage equilibrium”. Nonrandom associations (“linkage disequilibria”) can be generated by a variety of evolutionary processes; their observation gives an indirect method for quantifying the rates of these processes. Thus, there has been considerable interest in estimating the strength of linkage disequilibria, motivated by varied aims: finding gene order and recombination rates (Hill & Weir, 1994); detecting genes responsible for human disease (Kaplan et al., 1995); detecting selection for particular gene combinations (Langley, 1977); and measuring patterns and rates of migration (Asmussen et al., 1987; Barton & Gale, 1993). This paper is concerned with understanding hybrid populations, in which the mixing of genetically distinct taxa can maintain strong associations even between unlinked genes (Li & Nei, 1974). Methods for estimating and interpreting pairwise associations are well worked out, both for nuclear–nuclear and nuclear–cytoplasmic associations (Hill, 1974a; Asmussen et al., 1987). However, data are usually available from several marker loci: here, we set out methods for combining data across loci, paying particular attention to the case where there are strong associations both between genes from the same gamete, and between genes inherited from different parents.
There are three key difficulties. First, estimates of associations between different pairs of loci are not independent of each other, especially when associations are strong. Secondly, we do not usually wish to estimate individual associations; in any case, there are far too many such associations with even a few loci. Rather, we wish to find some composite measure which gives information about the process responsible for generating linkage disequilibrium. (For example, random drift and recombination will generate a distribution whose parameters might be estimated from the variance in pairwise disequilibrium; e.g. Langley, 1977.) Thirdly, unless samples are of zygotes freshly generated by random mating, there will be associations between genes from different parents; with diploids, these cannot be distinguished from associations between genes derived from the same gamete.
Most of the literature on estimating linkage disequilibrium has been concerned with the third problem, of resolving genotypes that are confounded in diploid data (for example, using the EM algorithm; Hill, 1974a,1975; Dempster et al., 1977; Long et al., 1995; Slatkin & Excoffier, 1996). There is also a good understanding of the distribution of associations generated by random drift within a single population (Hill, 1974b, c). The primary motivation for this study is the estimation of associations generated by the mixing of populations. This raises particular difficulties, because there are often strong associations, both within and between genomes; it is also particularly simple, in that admixture models make straightforward predictions as to the structure of associations in a population.
The crucial requirement is for some model which specifies the frequencies of genotypes in terms of a reasonably small number of parameters. Given such a model, the parameters can be estimated by maximum likelihood. Such a model is also necessary for other statistical problems where genotype frequencies must be specified: for example, estimating paternities or mapping quantitative trait loci. Here, we show that many kinds of admixture lead to a model with a simple structure, and show how the parameters of this model can be estimated by maximum likelihood. First, however, we justify the choice of this model by discussing alternative methods which might be used to determine the structure of genotype frequencies.
The key difficulties arise because of the very large number of possible genotypes. For example, with two alleles at each of five loci, there are 25=32 haplotypes and 35=243 distinguishable diploid genotypes. Unless samples are extremely large, one cannot estimate even haplotype frequencies. Moreover, if gametes are not combined at random, the genetic structure is determined by the much larger number of diploid genotypes; yet, these cannot be distinguished without knowing which gene came from which parent. The very large number of genotypes rules out the most obvious method: to make a maximum likelihood estimate (MLE) of the genotype frequencies. Even in large samples, most diplotypes, and even most haplotypes, may be missing. Therefore, the MLE would be that only observed genotypes are actually present, implying complete associations between certain gene combinations. This can raise serious difficulties. For example, suppose that we wish to find whether some family was sired by a known male, or by some unknown father. If we suppose that genotypes that are absent in our sample are absent in the rest of the population, then there will be an undue tendency to assign paternity to implausible parental genotypes which happen to be represented amongst the known individuals.
The problem, therefore, is to describe the genotype frequencies in terms of a small number of parameters; the description must be simple enough to make calculations tractable, and must include the structure of the actual populations under study. A popular solution to this kind of problem has been to use the method of “maximum entropy” (Guiasu & Shenitzer, 1985; Phipps & Brill, 1995). The entropy of a sample is defined as S=ΣXg[X ] log[g[X ]], where g[X ] is the frequency of genotype X. Given the constraint ΣXg[X ]=1, entropy is maximized when all genotypes are equally common. Given further constraints on allele frequencies, the maximum entropy is at linkage equilibrium. With constraints on pairwise linkage disequilibria, the maximum entropy solution defines the genotype frequencies in terms of the small number of pairwise coefficients. In the haploid case, the solution is g[X]=exp[Σi λiXi + Σi,j λi, jXiXj]/Z, where Z is a normalizing constant, and Xi=0 or 1 defines the state of the ith locus. The λs are determined by the equations ∂ln(Z)/∂λi=pi, ∂ln(Z)/∂λi,j=pipj + Ci,j, where pi is the allele frequency, and Ci,j is the covariance (equivalent to the linkage disequilibrium) between loci i and j. This method has a tempting generality, but suffers two drawbacks. First, numerical solutions involve complicated transcendental equations, which become intractable in the diploid case. Secondly, there is no reason why evolutionary processes should lead populations to have “maximum entropy”. The method has been justified as reflecting our ignorance of unknown frequencies (analogous to arguments justifying parsimony in phylogeny reconstruction; Sober, 1983). However, even though we may not know actual genotype frequencies, we may know the patterns of genotype frequencies likely to be produced by evolution: estimation procedures should be based on these patterns. Moreover, the parameter estimates are not interesting in themselves: they can only be interpreted in the light of some evolutionary model.
A related approach is that taken by Haber (1984) where a log-linear model is used to parameterize genotype frequencies. For two loci (labelled i, j), the frequency of the diploid combination {{Pi, Pj},{Pi*, Pj*}} (where i and j label the alleles inherited from the mother, and i*, j* those from the father), is given as exp[μ + αi + αj + αi* + αj* + βij + βi*j* + βii* + βjj* + βij* + βji*]. Only first- and second-order interactions are considered, but the method could be extended to include higher-order interactions and multiple loci. A difficulty with this method is that, in order to determine the parameters uniquely, some arbitrary reference point needs to be assigned, which will not necessarily have a biological interpretation. For example, Haber (1984) suggests setting the sums over the indices to zero. With no higher-order interactions, the allele frequencies for locus i are then p1=exp[μ + α1] and p2=exp[μ − α1]; however, the parameters μ, α do not bear a simple relation to allele frequencies in the presence of interactions. Another problem is that the parameters of the log-linear model measure net effects; the parameter βjj*, which measures deviations from random mating with respect to locus j, could be zero, but if some of the other five sets of two-way parameters, β, are nonzero, the covariance between alleles at locus j will not be zero. Finally, there is still the problem of estimating a large number of parameters; simply setting parameters for higher-order interactions to zero may cause erroneous estimates of lower-order interactions.
Here, we suppose that the population is subject to continuing immigration by adults, from two source populations with different allele frequencies. Mixing of divergent populations builds up associations between genes inherited from the same parent, and from different parents. In each generation, associations between genes inherited from different parents are eliminated by segregation and recombination, and then built up again by immigration. We assume that continued migration does not dissipate differences between populations; in reality, these may decay, or may be maintained by selection. However, the assumption of a short-term balance between recombination and migration is accurate even when allele frequencies are homogenizing, or when selection maintains them in the face of gene flow (Barton & Gale, 1993; Kruuk, 1997; Barton & Shpak, 2000a; Kruuk et al., 1999).
It would be possible to fit a specific model of admixture, giving estimates of migration rates, allele frequencies in the source populations, and so on. However, the distribution of genotype frequencies depends on the detailed migration pattern, which is often unknown. In particular, migration at a low rate from divergent populations maintains a small fraction of genetically distinct individuals; a higher rate of immigration from less divergent populations may maintain the same genetic variance, but much smaller higher-order associations. We therefore aim to find a genetic structure which covers this range of admixture models; estimates of this general structure can then be compared with particular migration models.
This approach is closely related to the simple practice of classifying hybrid individuals into various classes of cross (Arnold, 1997). If hybridization is rare or recent, then there may be distinct classes of parental and F1 genotypes. Backcrosses can be identified because they are homozygous only for genes from a single taxon; given enough markers, the number of generations of backcrossing could be estimated (Goodman et al., 1999). However, if hybridization has been extensive, individuals can no longer be unambiguously assigned (at least, without an inordinate number of markers). The classification can then be misleading: for example, putatively F1 genotypes may be more likely to be generated by chance than by an actual cross between parental genotypes. One could still describe a population as some mixture of parentals, F1s, backcrosses and F2s. However, this mix is not uniquely determined by genotype frequencies. We discuss below the relation between our description of genotypic structure, and classification by hybrid status.
The analysis falls into three sections. First, it is shown that models of admixture, involving both short- and long-range migration, lead to associations among genes which depend on the number of genes involved, and on how many of each kind come from each parent. Associations involving particular loci are proportional to the divergence in allele frequency for that locus; this makes it simple to rescale, so as to allow for variation in associations across loci. Secondly, an algorithm is presented for making maximum likelihood estimates of the within- and between-genome associations, and for testing hypotheses as to their magnitude. Finally, the accuracy of this algorithm is demonstrated against simulated datasets. Procedures for analysing multilocus admixture models, and for maximum likelihood estimation, are implemented in MATHEMATICA 3.0, and are available from http://helios.bto.ed.ac.uk/evolgen/.
Models of admixture
Suppose that proportions m1, m2 migrate in from two source demes, with allele frequencies p1;i, p2;i at locus i. At equilibrium, the allele frequency in the deme of interest is pi=(m1p1;i+m2p2;i)/(m1+m2). The notation can therefore be simplified by letting p1;i=pi − Uδpi, p2;i=pi + Vδpi, where the migration rates are m1= mU, m2=mV, and where δpi=(p2;i − p1;i). Thus, m is the total immigration rate, and U, V are the proportions of migrants from each source (U+V=1). Note that U is determined by the ratio of migration rates, and so is the same for all loci; it is not necessarily equal to the mean of the pi. If alternative alleles are fixed in the source demes, then δpi=1, and pi=U. However, if δpi < 1, then the equilibrium allele frequencies pi may vary across loci.
We must now determine the associations among loci at equilibrium. Following Turelli & Barton (1994), we define these as multivariate cumulants across sets of genes. Consider loci with two alleles, labelled by Xi=0 or 1; in a diploid, the copies inherited from the mother and father are labelled Xi, Xi*, respectively, and the full genotype is defined by the pair of vectors {X, X*}. Deviations from the population mean are defined by ζi=(Xi − pi), where pi=E[Xi]. The pairwise association between genes at i and j, inherited from the mother, can be described by the covariance between Xi, Xj, denoted C{i,j} ≡ E[ζiζj]. Similarly, the pairwise association between the gene at i inherited from the mother, and the gene at locus j inherited from the father, can be described by C{i,j*}≡E[ζiζj*], where pi*=E[Xi*]. (The coefficient C{i,i*} describes the deficit of heterozygotes at locus i). Higher-order associations are described in a similar way to multilocus moments (Barton & Turelli, 1991.) The genotype frequencies can be reconstructed as a linear combination of the various multilocus moments. With two loci, for example, the frequency of the double homozygote {{1, 1}, {1, 1}} is:

The frequency of other genotypes is found by replacing pi by qi, and changing the sign of every coefficient CU, whenever an allele Xi=1 is replaced by Xi=0 in (1). Thus, the 16 genotype frequencies are determined by four allele frequencies, six pairwise associations, four three-way associations, and one four-way association. The frequencies of more complex genotypes could be des- cribed in a similar way, by use of higher-order moments.
The population can also be described in terms of multilocus cumulants rather than moments (Turelli & Barton, 1994). Cumulants are polynomial functions of the moments; the second- and third-order cumulants are identical to the central moments, but the fourth- and higher-order moments differ. For example, κ{i,j,k,l}= C{i,j,k,l,} − C{i,j}C{k,l} − C{i,k}C{j,l} − C{i,l}C{j,k}. Cumulants can be thought of as describing the association amongst a set of genes, over and above those expected from the lower-order associations amongst the various subsets. They are defined so as to be additive. Thus, if an additive trait is constructed as the sum of contributions of all the genes (∑i(Xi+Xi*)), then its kth cumulant is the sum of all cumulants amongst sets of k genes. We choose cumulants to describe admixture models because higher-order cumulants are small in admixture models; all cumulants of the same kind are of similar magnitude (see below); and at Hardy–Weinberg, all cumulants involving genes inherited from different parents are zero. (This is not the case for moments: for example, C{i,j,k*,l*}=C{i,j}C{k*,l*} for a population in Hardy–Weinberg proportions.)
Genotype frequencies are readily derived from eqn 1 by expressing moments in terms of cumulants. For example:

The same rules apply for finding the frequency of other genotypes as above.
These expressions can readily be extended to allow for multiple alleles, provided that there are ultimately just two sources of migrants, which are in linkage equilibrium. Suppose that within deme 2, allele αi at locus i is at frequency similarly for deme 1. Now, regardless of their actual allelic state, label alleles that derive from either of the two demes as Xi=0 or 1. Then, the frequency of an individual entirely composed of alleles derived from deme 2 is given by eqns 1 or 2; and the chance that this individual carries alleles αi is the product of the s. The net frequency of some allelic combination is given by a sum over all origins of those alleles, multiplied by their probability, given that origin. This sum will simplify if certain alleles are found only in one or other source.
The algorithms which define the effects of recombination and random mating within demes are given in Turelli & Barton (1994); migration simply involves a linear mixture of moments. The algorithms can be found at http://helios.bto.ed.ac.uk/evolgen/. Here, we apply them to find the associations which would be found at equilibrium, in a balance between migration and recombination. We assume no linkage between loci, which is the case of most practical interest. (Linkage introduces considerable difficulties, because associations will decrease in a com- plicated way with recombination rate.) The expressions below assume symmetry across the two sexes; they also apply to cytonuclear disequilibria, provided that evolu- tionary processes apply equally to both sexes.
Below, we give the equilibrium solutions for two extreme cases. First, migrants might come from source populations which are in linkage equilibrium. This will necessarily apply if the sources are fixed for alternative alleles, and so we refer to this case as “long-range migration”. Secondly, migrants might come from neighbouring demes with different allele frequencies, but with the same levels of association. This is an approximation to a stepping-stone model, where migration is between neighbours which are in a similar state. It ignores the diffusion of linkage disequilibrium, which tends to reduce associations in the centre, and increase them at the edge. (This is the “quasi-linkage equilibrium” approximation of Barton (1986) and Kruuk et al. (1999). We compare the “short-range migration” approximation with exact simulations below.
Assuming symmetry across the sexes, pairwise associations under these two approximations are:

Associations between the two copies of an allele at the same locus, κ{i,i*}, which describe heterozygote deficit, are given by replacing j* by i* in the expression for κ{i,j*} above. The influx of linkage disequilibrium from the source populations assumed under the model of short-range migration increases associations within genomes by a factor (1+m)/(1 − m/3), but does not affect associations between genomes. With both kinds of migration, the associations between genomes are smaller because they are broken down in every generation by segregation. All the expressions in eqn 3 are for associations measured immediately after dispersal; among zygotes, there will be no associations between maternal and paternal genomes. Associations within genomes will be reduced to κ{i,j}*= (κ{i,j}+κ{i,j*})/2 by random segregation and mating.
Three-way associations are given by similar expressions:

Finally, we give the four-way associations:


Expressions for higher-order associations become more complicated, but share a similar structure. In particular, all associations involving a set of loci U are given by the product Πi∈Uδpi, multiplied by a factor independent of the loci involved. This is important, because it gives a simple way of scaling out variation across loci, and because it allows associations between genes at different loci, κ{i,j*}, which are not directly observable, to be estimated from observations on heterozygote deficit, κ{i,i*}. It also allows the effects of immigration from near and far to be distinguished: if δp is small, then associations among many genes become small, whereas if δp is close to 1 (its maximum value), then all higher-order associations are proportional to mUV, and may be large. This is reflected in a leptokurtic distribution of the number of alleles from one or other taxon.
The strength of associations generated by admixture does not depend directly on allele frequency. It is proportional to mUV, which is the harmonic mean of the immigration rates mU, mV. If δpi < 1 (for example, with short-range migration), then allele frequencies might vary across loci without directly affecting the associations between them.
Associations between genes inherited from different parents depend primarily on the numbers of genes involved. For example, eqn 5 shows that κ{i,j,k,l*}, κ{i,j*,k*,l*} are identical to κ{i,j,k*,l*} for short-range migration, and with long-range migration, are extremely close over the whole range of migration rates (Figs 1 and 2). Similar calculations show that all fifth- and sixth-order cross-genome cumulants are similar; agreement becomes closer as migration rates become asymmetric (i.e. U ≪ 1/2 or ≫ 1/2). This similarity between different classes of association arises because higher-order associations are almost entirely eliminated by segregation: the nth-order association decreases by a factor 2n−1 every generation. Therefore, associations are close to those built up by admixture in the current generation, which is the same for all nth-order associations.

Comparisons between the three kinds of four-way association, κ{i,j,k*,l*}, κ{i,j,k,l*}, κ{i,j,k,l }, (bottom to top) plotted against migration rate, m; U=0.1. Values are from eqn 5, scaled relative to δpi δpj δpk δpl. (a) Long-range migration, (b) short-range migration. With short-range migration, (b), κ{i,j,k*,l*} is identical to κ{i,j,k,l*}. Note that with short-range migration in a stepping-stone model, (b), m < 1/2, and δp ≪ 1; therefore, the actual associations may be much smaller than with long-range migration, even though the scaled values shown here are larger.

Comparisons between the three kinds of four-way association, κ{i,j,k*,l*},κ{i,j,k,l*},κ{i,j,k,l} (bottom to top at p=0.1), plotted against allele frequency, U; m=1/2. Values are from eqn 5, scaled relative to δpi δpj δpk δpl. (a) Long-range migration, (b) short-range migration. With short-range migration, (b), κ{i,j,k*,l*} is identical to κ{i,j,k,l*}.
Calculating genotype frequencies
In admixture models with random mating and with just two sources of immigrants, genotype frequencies can be specified in terms of allele frequencies, pi ; relative divergence across loci, δpi; Kth-order within-genome associations, κ0,K; and between-genome associations involving J genes from one parent, and K from the other, κJ,K. With n loci, and symmetry across the sexes, there are (n − 1) within-genome coefficients; n(n − 1)/2 between-genome coefficients, κJ,K; n allele frequencies, pi; and (n − 1) divergences, δpi. Therefore, the 3n genotype frequencies are determined by (n2+5n − 4)/2 parameters. (Note that the δpi only give the relative importance of each locus, and could be multiplied by an arbitrary constant; they are therefore associated with (n − 1) degrees of freedom.) For example, with four loci, the 81 distinguishable diploid genotypes are specified by just 16 parameters; with five loci, the 243 distinguishable genotypes depend on 23 parameters. The similarity between all nth-order cross-genome associations (Figs 1 and 2) suggests a further simplification: to equate all cross-genome cumulants involving the same number of loci. However, this can lead to negative genotype frequencies: even the small changes needed to force equality among cross-genome associations of the same order can force genotype frequencies outside their valid range.
If the set of genes U all come from the same parent, κU=κ0,|U| (Πi∈Uδpi), where |U| is the number of genes in U; if a set U come from one parent, and U* from the other, then κU,U*=κ|U|,|U*| (Πi∈Uδpi)(Πi∈U*δpi*). The genotype frequency (1) can then be written compactly as follows:

where Γ=( pipj ( piδpj + δpi pj)δpi δpj),

where CJ,K represents the multilocus moment, and κJ,K the multilocus cumulant.
The frequency of other genotypes is found by replacing pi by qi and δpi by −δpi whenever an allele Xi=1 is replaced by Xi=0 in (1). In general, with n loci, C is the matrix of scaled moments (with C1,0 ≡ 0, C0,0 ≡ 1), and Γ is a vector indexed 0 ,..., n, whose ith element is the sum of products of (n − i ) distinct allele frequencies and the remaining i distinct δps. The complete matrix of 2n × 2n diploid genotypes can be written as T · C · T*T, where T is the 2n × (n + 1) matrix, each row being a permutation of the vector Γ defined in (6). For example, with two loci:

Expressed in this form, genotype frequencies can be calculated efficiently, since T depends only on allele frequencies, and C depends only on the moments or cumulants. Thus, when exploring the statistical fit of alternative moments, T need not be recalculated.
The model discussed so far has assumed migration from just two source demes. More complex models, with migration from several demes, will retain the same structure only under special conditions. The linkage disequilibrium after migration is a linear sum of the disequilibria contributed by each source. Therefore, the effects of each locus will scale by a factor δpi only if the deviations in allele frequency of all the sources from the target deme are proportional to this quantity. That is plausible if there is mixing between two taxa, via some network of demes, with the δpi corresponding to the difference in allele frequency at locus i between the parental taxa. For example, consider a cline. If there is diffusion from neighbouring demes, plus a lower influx from the parental taxa, then associations will scale with δpi provided that the gradient of the cline at some locus is proportional to the divergence between the taxa — which will be the case for neutral models. In general, however, admixture between large numbers of demes could produce any distribution of genotype frequencies.
A simple method for estimating multilocus moments
Suppose that we wish to estimate the pairwise linkage disequilibria, C0,2, C1,1 from a set of genotype frequencies; assume for the moment that the divergences are δpi=1. The coefficient C0,2 is the usual measure of pairwise linkage disequilibrium between genes derived from the same gamete, whereas C1,1 is the measure of the deficit of heterozygotes. One possibility would be to use the variance of an additive trait, z≡∑i(Xi+Xi*), where X is the vector giving the states of each gamete. This variance is just var(z)= ∑i∑j(C{i,j}+ C{i,j*}+C{i*,j}+C{i*,j*})=2(∑ipiqi+n(n−1)C0,2+n2 ×C1,1), since C{i,i}=piqi. The associations within and between genomes can be disentangled by estimating C{i,i*} from the deficit of heterozygotes at individual loci. Because we assume that between-genome associations are the same whether they involve the same or different loci (C{i,i*}=C{i,j*}=C1,1), this allows the component of var(z) attributable to cross-genome associations to be separated from that attributable to associations within genomes (see Barton & Gale, 1993; Kruuk, 1997).
This approach extends to give a simple way of estimating the whole matrix of moments, Cj,k, from a set of 3n diploid genotype frequencies. Each locus is described by the value Xi+Xi*=0, 1 or 2. However, the underlying genotypes {0, 1} and {1, 0} cannot be distinguished in heterozygotes: this makes it impossible to estimate directly all the multilocus moments. Nevertheless, provided that allele frequencies are known, and are the same amongst male and female gametes, two useful quantities can be defined for each locus. First, the additive effect is given by zi≡ζi+ζi*=Xi+Xi*−2pi. Secondly, the deviation from Hardy–Weinberg proportions is described by φi≡ζiζi*, which takes values {pi2,−piqi,qi2} for Xi+Xi*={0,1,2}. Next, find the average value Ma,b of all those terms which contain a factors zi, and b factors φi, all with distinct indices: Ma,b≡〈Πi∈A(ζi+ζi*) Πj∈B(ζjζj*)〉, where A, B are sets of distinct and nonoverlapping indices, and < > indicates an average over all (n!/a!b!(n−a−b)!) such terms. (For example, with four loci, M2,2=(z1z2 φ3φ4+z1 z3 φ2φ4+⋯)/6.) If the genotype frequencies have the form given by the admixture model, with δp=1, then the expectation of Ma,b is
 (Note that because the Ma,b have been defined to include only terms with distinct indices, moments such as C{i,i}=pi qi do not arise; the expression for E[Ma,b] therefore does not include allele frequencies.)
(Note that because the Ma,b have been defined to include only terms with distinct indices, moments such as C{i,i}=pi qi do not arise; the expression for E[Ma,b] therefore does not include allele frequencies.)
Taking all possible Ma,b provides a set of linear equations which uniquely determine all possible Cj,k. For example, for three loci, the moments Cj,k are given in terms of the expectations of Ma,b as follows:

It would be straightforward to find unbiased estimators for the Ma,b, based on small samples. However, because the degree of bias is negligible compared with the standard deviation of the estimates, the additional complication is not warranted.
If the δpi vary across loci, the same method can be used, provided that zi is scaled relative to δpi, and φi relative to δpi2. Then, Ma,b≡Πi∈A(ζi+ζi*)/δpi Πj∈B(ζjζj*)/δpi2. Clearly, this method fails if any of the δpi are zero. In itself, this poses no difficulty, because such irrelevant loci could be deleted. However, the high weight contributed by loci with weak associations (δp ≪ 1) suggests that the method may be statistically inefficient if there is wide variation in the δpi.
This method gives satisfactory estimates of individual coefficients. However, it is unsatisfactory as an estimator of the overall genotypic structure, described by the full set of moments, because individual genotype frequencies may be negative. Even if a population has the structure required by the admixture model, samples from that population will vary from that structure, and estimates of allele frequencies and moments made using eqn 8 may not give valid genotype frequencies.
Maximum likelihood estimation
Expressions for the genotype frequencies are linear combinations of the multilocus moments. The log likelihood (log(L)) of the parameters (pi, δpi, κj,k) is given by summing log(g[X ]) over all observed genotypes, X. The maximum likelihood estimate (MLE) is found by maximizing log(L), subject to the constraint that all g[X ] must be non-negative, including those genotypes which did not happen to arise in the sample. This is a nontrivial task, because a very large number of constraints are involved. It can be simplified in two ways. First, allele frequencies can be set to their observed values; this considerably speeds the calculation, because calculating the matrix T (a function of the allele frequencies) is otherwise the limiting step. The observed allele frequencies are not quite the same as their MLE in the presence of linkage disequilibrium; indeed, with moderate linkage disequilibrium (e.g. with m > 0.1 below), the sampled allele frequencies may be incompatible with the true Cs that generated the sample, leading to negative genotype frequencies. It will therefore be necessary to fit allele frequencies as well as linkage disequilibria in the simulations below, because we wish to compare with those true values. However, that may not be necessary in making estimates from real data.
Secondly, the δpi may not need to be estimated. They may be known a priori, from knowledge of the source demes. If they are not, then they can be chosen so as to fit the average pairwise association for each locus. The covariance between the diploid genotype (scored as 0, 1 or 2) at loci i and j is cov[Xi+Xi*,Xj+Xj*]≡ Ĉi,j=2(C{i,j}+C{i,j*}), and must equal αδpiδpj, where α depends on migration rates, etc. Therefore, the sum of the covariances involving locus i is:

Given some α, eqn 9 determines the δpi which will fit the observed Ĉi,•. The choice of α is arbitrary, because the δpi can be increased, and the CJ,K decreased, so as to leave the predicted linkage disequilibria and genotype frequencies unaltered. One possibility is to suppose that the migration rates are in proportions U:V equal to the mean allele frequencies in the sample, and then take the largest δpi which still allow valid allele frequencies in the hypothetical source populations (i.e. 0 <pi − Uδpi, 1 > pi + Vδpi). Heterogeneity across loci can be tested by comparing the likelihood of this choice with setting δpi=1.
Once the pi and δpi are chosen, the likelihood must be maximized with respect to the κJ,K, subject to constraints on genotype frequencies. For populations close to Hardy–Weinberg and linkage equilibrium, simple Newton–Raphson maximization is adequate. However, when linkage disequilibria are strong, this often leads to solutions with negative frequencies of unobserved genotypes. We therefore deal with such cases using the Metropolis algorithm (Metropolis et al., 1954). A random change is made to parameter k; this is drawn from a symmetrical uniform distribution, with maximum value ±Δk. If the new parameter set is valid, and if it increases the likelihood, it is accepted. If it is valid, but decreases the likelihood by a factor θ < 1, then it is accepted with probability θ1/T. The size of random perturbations is optimized by increasing Δk slightly if a change in parameter k is accepted, and decreasing it if the change is rejected. This procedure, applied to each of the parameters in turn, generates a random walk with a probability distribution L1/T. The parameter T determines how closely this distribution clusters around the optimum (or optima); it is analogous to a temperature, in that with large values of T, the system wanders randomly over a wider range, whereas with small T, it “freezes” to some local optimum. The global optimum can be found by starting at some high T, and gradually cooling to T=0 (“simulated annealing”; Kirkpatrick et al., 1983). Setting T=1 gives a distribution proportional to the likelihood, which can be used to generate support limits on the parameters.
Likelihood provides a natural way of comparing nested hypotheses (Edwards, 1972; Mangel & Hilborn, 1996). One can find in turn the likelihood that the population is in Hardy–Weinberg and linkage equilibrium; that gametes are combined at random, but that there are associations between genes derived from the same gamete, κ0,2; that there are also higher-order associations within gametes, κ0,K (K > 2); that there are pairwise associations between genes inherited from different parents, κ1,1; and finally, that all associations within and between genomes contribute. One might also compare the likelihood that all allele frequencies, pi, are equal, or that the contribution of each locus to linkage disequilibrium, δpi, is the same. In order to assess the relative plausibility, one must trade an increase in likelihood against the number of parameters fitted. One approach is to treat the increase in log likelihood obtained by fitting ν parameters as a statistic, which approaches a ½χ2ν distribution in large samples. However, this asymptotic result may not be accurate when samples are small, and when estimates are bounded by constraints. It would be possible (though extremely tedious) to find the exact distribution of the likelihood ratio statistic by simulation, or by bootstrap resampling. An alternative approach treats the likelihood itself as the criterion for inference; a plot of log likelihood against the parameters gives a measure of their relative plausibility. A difference in log likelihood of 2 units, which corresponds to one hypothesis being e2 = 7.4 times as likely as the other, can be used as a conventional threshold for acceptance. When hypotheses differ by several degrees of freedom, it is convenient to take thresholds from the ½χ2ν distribution, without applying a significance test as such. An alternative procedure is the Akioke information criterion (Mangel & Hilborn, 1996), under which a model is to be preferred if it has a larger value of log(L) − 2ν, which trades each degree of freedom against 1/2 a unit of log(L). Finally, one could use the Metropolis algorithm, with T=1, to generate a random walk proportional to the likelihood. The marginal distribution of each parameter then gives its likelihood, weighting the other parameters by their likelihood. This procedure amounts to Bayesian inference with uniform prior. The method described here is thus compatible with varied statistical philosophies.
We illustrate this approach using data on the genotypes of 37 toads sampled from the hybrid zone between Bombina bombina and B. variegata in Croatia (sample 1063 from MacCallum et al., 1998). The toads were scored for four unlinked and diagnostic allozymes (IDH, AK, MDH and LDH). The numbers of each genotype observed are given in Table 1, and are compared with those expected under several hypotheses. The log likelihood that the population is in Hardy–Weinberg and linkage equilibrium is −45.27. On the assumption that all loci are equivalent (δpi=1), a significant improvement is achieved by allowing pairwise associations within genomes (log(L)=−36.69; κ0,2=0.035). There is a further significant gain in allowing pairwise associations between genomes, representing a deficit of heterozygotes (log(L)=−34.12; κ0,2= 0.015, κ1,1=0.035). There is little further gain in fitting all the other higher-order associations (log(L)=−30.15, giving an increase in log(L) of 3.97 for an additional 11 d.f.). Allowing associations to vary across loci, by fitting δpi, gives a marginal improvement (log(L)=−31.31 with κ0,2= 0.021, κ1,1= 0.013; δp={0.37, 1.12, 1.18, 2.03}). This increase in log (likelihood) of 2.81 is not significant when compared with the asymptotic ½χ23 distribution (P=13%). Figure 3 shows how the likelihood depends on κ0,2 and κ1,1. The most accurate estimate is of the net covariance between loci (κ0,2+κ1,1); this is reflected in the relative closeness of the contours as both κ0,2, κ1,1 increase to top right. The hypothesis that κ1,1= 0 can be rejected: the contour corresponding to 2 units of log(likelihood) (second down from the peak) does not cross the horizontal axis (see Table 1). However, the hypothesis that κ0,2=0 cannot be rejected: that is, gametes might be in linkage equililbrium, provided that there is a strong enough heterozygote deficit.

The log likelihood surface for data from site 1063, plotted as a function of pairwise within- and between-genome association, κ0,2, κ1,1. All other associations are set to zero; δpi are fixed at 1, and allele frequencies are fitted by maximum likelihood. Contours of log likelihood are drawn at unit intervals.
Testing against simulated datasets
In order to test these estimation procedures, samples of 100 individuals were taken from a population subject to immigration from two source demes, fixed for alternate alleles (“long-range migration”). Samples were taken after migration, at which point the population would be in linkage disequilibrium and would deviate from Hardy–Weinberg. Table 2 shows the results of fitting various models to 100 replicate datasets, assuming two unlinked loci at U=0.2 or V=0.5, and total migration rates m=0, 0.1, 0.2, 0.3. (Note that with just two loci, the value of δpi is confounded with the values of the coefficients, and so may arbitrarily be set to 1.) For this problem, convergence of the Metropolis algorithm seems relatively fast. In all cases, parameters were changed 400 times each at T=2, 1 and then 200 times each at T=0.5, 0. Repeating this algorithm usually gave values of log(L) which differed by less than 0.1. In each case, allele frequencies were fitted by maximum likelihood. Initially, allele frequencies were set at their sampled values; if that did not give positive genotype frequencies, then allele frequencies were set equal, at a value which would give positive frequencies. This method sometimes failed to find a valid starting point (see below).
Maximum likelihood estimation of all the cumulants (second row of each table) gave results close to the true values; there is little bias, through κ1,1 is slightly underestimated for U=0.5, m=0.2, 0.3. The standard deviation of estimates of κ0,2 averages 0.0252 for U=0.5, and 0.0194 for U=0.2; within-genome associations can thus be detected reliably for low migration rates (m=0.1). The standard deviation of estimates of κ1,1 averages 0.0182 for U=0.5, and 0.0156 for U=0.2; between-genome associations can therefore be reliably detected for m=0.2 at U=0.2 and 0.5. The standard deviation is expected to scale approximately with the square root of sample size.
The simple method of eqn 8 performs comparably with maximum likelihood estimation: standard deviations are similar, and again, the only evidence of bias is a slight underestimation of κ1,1 for p = 0.5 and large m. However, this method does not provide a general method of hypothesis testing, and may not give valid predictions of genotype frequencies: the cumulants estimated in this way, combined with the sampled allele frequencies, often lead to negative genotype frequencies.
Throughout, the distribution of log(L) fits reasonably well with the expected ½χ2ν distribution, which has mean and variance ν/2 (where ν is the number of residual degrees of freedom). For example, one can compare the log likelihood, fitting all coefficients (second row), with that obtained by setting coefficients equal to their true values (fifth row). The difference in mean log likelihood is close to the expected value of 2 (e.g m=0.3, p=0.2; Δlog(L)=1.82). However, in a few cases (all for p=0.2), fitting more parameters apparently led to a lower likelihood. This occurs when the Metropolis algorithm fails to find the global optimum; in principle, the difficulty could be solved by cooling more slowly from a higher temperature. This problem only arose in 2/800 cases when the likelihoods of the true values of all the cumulants were compared with the likelihood, fitting all the cumulants (fifth vs. second rows in Table 2). The problem was more frequent (30/800 cases) when the likelihood of the pairwise cumulants (κ1,1, κ0,2) was compared with the likelihood, fitting all cumulants (fourth vs. second rows in Table 2). This is because here, both likelihoods involve stochastic optimization using the Metropolis algorithm.
Hardy–Weinberg and linkage equilibrium was rejected in all cases for m > 0.1; in three cases for m=0, U=0.5; in 79 cases for m=0.1, U=0.5; and in four, 74 cases for m = 0, 0.1, U=0.2 (second vs. first rows; 2 d.f.). Thus, where the population was in fact in Hardy–Weinberg and linkage equilibrium (m=0), the true hypothesis was rejected ~5% of the time, whereas for m ≥ 0.1, Hardy–Weinberg and linkage equilibrium was rejected in most cases.
The number of replicates in which the true values of the cumulants were rejected, at the 5% level, was close to expectation. The hypothesis that the pairwise associations κ1,1, κ0,2 equal their true values was rejected in 7, 5, 8, 5 cases for m=0, 0.1, 0.2, 0.3 and U=0.5, and 3, 8, 5, 3 cases for U=0.2 (second vs. fourth rows; 2 d.f.). The hypothesis that the all cumulants equal their true values was rejected in 3, 5, 7, 5 cases for m=0, 0.1, 0.2, 0.3 and U=0.5, and 4, 5, 3, 4 cases for U=0.2 (second vs. fifth rows; 2 d.f.). This agreement is some what surprising, given that the numbers of most genotypes are small, and the maximum likelihood estimate is often bounded by constraints on genotype frequencies.
Table 3 shows results of simulations with four unlinked loci; the migration rate is m=0 or 0.3, and U=0.2 or 0.5. As before, allele frequencies are fitted by maximum likelihood, and all loci are equivalent (i.e. δp=1). (The case where pairwise associations were fixed at their true values and the remainder fitted is not now shown.) The simple method of eqn 8 (third row of Table 3) performs well, in that the mean of estimates of κ1,1, κ0,2 is close to the true value, and the standard deviation across replicates is similar to that of maximum likelihood estimates. However, the matrix of moments estimated using eqn 8 usually gives negative genotype frequencies when combined with sample allele frequencies; this reflects the strong constraints on the moments with four loci. With high migration (m=0.3), maximum likelihood estimates of κ1,1, κ0,2 are on average ~10% lower than the true values; with no migration, there is a small positive bias for κ0,2 (0.0136 for U = 0.2, 0.0094 for U=0.5); a small positive bias for κ1,1 with U=0.2 (+0.0041); and a small negative bias for κ1,1 with U = 0.5 (−0.0051).
Likelihoods now agree poorly with asymptotic theory; this is to be expected, as there are now stronger constraints on the parameters, and because the genotype frequencies are lower. The mean residual log(L) are now substantially smaller than the expected value, ν/2 (first column of Table 3). Comparison of hypotheses also shows smaller differences in log(likelihood) than expected for m=0.3. For example, with U=0.5, the improvement in log(likelihood) obtained by fitting 13 cumulants is on average 3.89, with variance 6.35; this compares with the asymptotic expectation of 6.5 for each. Examination of the cumulative distribution of Δlog(L) shows that it is shifted to the left by ~2.6 units for U=0.5, m=0.3, and ~4.2 units for U=0.2, m=0.3. Correspondingly, the true matrix of cumulants is rejected less often than expected from asymptotic theory: at P=5%, it is rejected 0/100, 1/100 times at U=0.2, 0.5 and m=0.3. For populations in linkage equilibrium (m=0), and U=0.5, agreement is better: the mean and variance of Δlog(L) are 5.55, 5.52, close to the expectation of 6.5. With U=0.2, the mean and variance of Δlog(L) are 0.93, 6.78; the mean is again lower than the expectation of 6.5. The true cumulants are rejected in 4/100 cases for U=0.5, and in 0/100 cases for U=0.2.
Discussion
If linkage disequilibria are generated by the mixing of two source populations, either directly or across a cline, they take a simple form: the magnitude of the association between a set of loci depends primarily on the number of each kind of allele derived from the maternal genome, and the number derived from the paternal genome (J, K, say). The state of the population can therefore be described by the allele frequencies, and by a matrix giving the various associations. These associations can be described either in terms of multilocus moments, Cj,k, or multilocus cumulants, κj,k ( j=0...n, k=0...n). There is a simple relation between the matrix of 22n diploid genotype frequencies, G, and the matrix of moments, C : G = Γ · C · ΓT, where Γ is a matrix which depends only on the allele frequencies (eqn 7). This method extends to allow for different allele frequencies in the male and female gamete pools, and for different degrees of divergence across loci.
Although the method described here is motivated by models of neutral admixture, it may apply to other cases in which different biallelic loci are equivalent: for example, where selection acts on an additive trait determined by unlinked loci. If all loci are interchangeable, such that all genotypes with the same numbers of ‘+’ alleles contributed by the maternal and paternal gametes (n,n*) are equally frequent, then the population can be described by the joint distribution of (n, n*). This symmetrical polygenic model has been investigated by Kondrashov (1984), Barton (1992), Doebeli (1996), Shpak & Kondrashov (1999) and Barton & Shpak (2000b); the diploid case has been treated by Kondrashov & Kondrashov (1999). This description is equivalent to that set out here in terms of a matrix of moments or cumulants. However, current theory is restricted to cases where loci are fully interchangeable. It may be that variation across loci (for example, in their effects on an additive trait) might be described in the same way as variation in the divergence, δp, in admixture models. It may also be that the full model could be approximated by a description in terms of pairwise moments or cumulants (c.f. Turelli & Barton, 1994). Such possibilities warrant further investigation.
The matrix of moments, CJ,K can be calculated as a linear combination of various products of additive and dominance effects associated with each locus (eqn 8). This method performs as well as maximum likelihood estimation, and is much faster; it also avoids the uncertainties of the Monte Carlo algorithm used here to maximize likelihood. However, this simple method does not allow the plausibility of different hypotheses to be compared, and more seriously, often leads to sets of allele frequencies and moments that predict negative genotype frequencies. In contrast, maximum likelihood estimation allows flexible comparison of nested hypotheses, and necessarily gives estimates consistent with positive genotype frequencies.
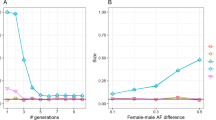
One motivation for this work was the need to combine information about pairwise linkage disequilibria across loci. When linkage disequilibria are weak, and samples are large, estimates of pairwise linkage disequilibrium should be approximately independent, even when pairs of loci overlap (e.g. E[C{i,j } C{i,k}]=0). The standard error of an estimate of the average pairwise disequilibrium should then decrease with the square root of the number of pairs of loci involved. However, with strong linkage disequilibria, estimates become strongly correlated, and so the standard error of estimates of pairwise associations should decrease more slowly with the number of loci. Figure 4 shows the standard deviation of estimates of κ1,1, κ0,2 as a function of m, for two and four loci, and for U=0.2, 0.5. At linkage equilibrium (m=0; left of Fig. 4), the standard error is on average smaller by a factor 0.57 with four loci, compared with two loci; this is somewhat higher than 1/√6=0.41, the factor expected if the six pairwise associations with four loci contribute independent information. In contrast, the standard error with m=0.3 (i.e. with strong linkage disequilibrium) is reduced only by a factor 0.84 as the number of loci increases from two to four.

The methods described here give estimates of associations of all orders. In principle, higher-order associations can give additional information: for example, short- and long-range migration can be distinguished by the rate at which associations decrease with the number of loci involved (see above). However, estimates of higher-order linkage disequilibria have high sampling errors. With two loci (Table 2), the third- and fourth-order cumulants κ1,2, κ2,2 are small for U=0.5, and so cannot be distinguished from zero with samples of 100 individuals. When allele frequencies are asymmetric (U=0.2), higher-order associations are stronger, and become detectable for m > 0.2 (Table 2). A more fundamental problem is that the values of higher-order associations are strongly constrained by the requirement that genotype frequencies be positive, and therefore depend on both allele frequencies and on pairwise associations. It therefore makes little sense to make separate estimates of associations of different orders. If possible, an evolutionary model described by parameters such as migration rates should be fitted.
Often, data from hybrid populations are analysed by classifying each individual on the basis of its genotype (see Arnold, 1997). Where hybridization is rare, so that only a few distinct classes of hybrid are present, this is appropriate. However, there are several difficulties with this approach. First, loci may not be diagnostic (i.e. fixed for alternative alleles in different taxa). Even a low level of polymorphism makes it difficult to distinguish between hybrids and parental genotypes. Secondly, even if loci are initially diagnostic, continued backcrossing into a population causes polymorphism to build up within the native gene pool, making it impossible to distinguish recent hybrids merely by the presence of marker alleles. Recent hybrids can be distinguished by the presence of multiple alleles typical of the immigrant population — in other words, by linkage disequilibrium between introgressing alleles (Goodman et al., 1999). If hybrids always cross with mates from a large native population, then individuals could in principle be classified by backcross generation. The kth backcross would carry a fraction 2−k of introgressed alleles; if this fraction is higher than the background level of polymorphism, and if enough loci are scored, then the backcross generation could be inferred. However, such assignment is inaccurate even with many loci (Boecklen & Howard, 1997), and is impossible when hybrids mate with each other to produce F2s or complex backcrosses. Because the proportion of backcross hybrids in a population increases with time, t, as 2t, matings between hybrids soon become likely.
An alternative approach which is often used is to classify individuals by the fraction of their alleles which derive from each parental taxon, rather than attempting to infer their detailed ancestry (Arnold, 1997). Although this is a convenient and simple way of summarizing multilocus data, the method may be misleading if loci are not strictly diagnostic. Even if marker loci are diagnostic, much information is lost. An improvement would be to classify individuals according to both the number of alleles derived from one reference taxon, and by the number of loci that are heterozygous. This is equivalent to a description in terms of a matrix of moments, estimated using eqn 8. However, eqn 8 is more flexible in that it can allow for nondiagnostic loci through variations in allele frequencies and divergences, δp.
The methods described here give a flexible framework for describing hybrid populations in terms of linkage disequilibria of various orders. The strengths of pairwise linkage disequilibria can then be used to infer quantities such as rates of gene flow and the degree of assortative meeting. The likelihood of the full set of genotype frequencies can be calculated, which allows more elaborate hypotheses to be addressed. For example, one could find the likelihood that offspring were sired by sampled individuals of known genotype, rather than by some farther from the unsampled population. These methods make possible a variety of statistical analyses of hybrid populations.
References
Arnold, M. (1997). Natural Hybridization and Introgression, Princeton University Press, Princeton, NJ.
Asmussen, M. A., Arnold, J. and Avise, J. C. (1987). Definition and properties of disequilibrium statistics for associations between nuclear and cytoplasmic genotypes. Genetics, 115: 755–768.
Barton, N. H. (1986). The effects of linkage and density-dependent regulation on gene flow. Heredity, 57: 415–426.
Barton, N. H. (1992). On the spread of new gene combinations in the third phase of Wright’s shifting balance. Evolution, 46: 551–557.
Barton, N. H. and Gale, K. S. (1993). Genetic analysis of hybrid zones. In: Harrison, R. G. (ed.) Hybrid Zones and the Evolutionary Process, pp. 13–45. Oxford University Press, Oxford.
Barton, N. H. and Shpak, M. (2000a). The effects of epistasis on the structure of hybrid zones. Genet Res, (in press).
Barton, N. H. and Shpak, M. (2000b). The stability of symmetrical polygenic models. Theor Pop Biol, (in press).
Barton, N. H. and Turelli, M. (1991). Natural and sexual selection on many loci. Genetics, 127: 229–255.
Boecklen, W. J. and Howard, D. J. (1997). Genetic analysis of hybrid zones: number of markers and power of resolution. Ecology, 78: 2611–2616.
Dempster, A. P., Laird, N. M. and Rubin, D. B. (1977). Maximum likelihood from incomplete data via the EM algorithm. J R Statist Soc B, 39: 1–38.
Doebeli, M. (1996). A quantitative genetic competition model for sympatric speciation. J Evol Biol, 9: 893–910.
Edwards, A. W. F. (1972). Likelihood. Cambridge University Press, Cambridge.
Goodman, S. J., Barton, N. H., Swanson, G., Abernethy, K. and Pemberton, J. M. (1999). Introgression through rare hybridisation: a genetic study of a hybrid zone between red and sika deer (genus Cervus), in Argyll, Scotland. Genetics, 152: 355–371.
Guiasu, S. and Shenitzer, A. (1985). The principle of maximum entropy. Math Intelligencer, 7: 42–48.
Haber, M. (1984). Log-linear models for linked loci. Biometrics, 40: 189–198.
Hill, W. G. (1974a). Estimation of linkage disequilibrium in randomly mating populations. Heredity, 33: 229–239.
Hill, W. G. (1974b). Disequilibria among several linked neutral genes in finite populations I. Mean change in disequilibria. Theor Pop Biol, 5: 366–392.
Hill, W. G. (1974c). Disequilibria among several linked neutral genes in finite populations II. Variances and covariances of disequilibria. Theor Pop Biol, 6: 143–148.
Hill, W. G. (1975). Tests for association of gene frequencies at several loci in random mating diploid populations. Biometrics, 51: 881–888.
Hill, W. G. and Weir, B. S. (1994). Maximum likelihood estimation of gene location by linkage disequilibrium. Am J Hum Genet, 54: 705–714.
Kaplan, N. L., Hill, W. G. and Weir, B. S. (1995). Likelihood methods for locating disease genes in nonequilibrium populations. Am J Hum Genet, 56: 18–32.
Kirkpatrick, S., Gelatt, C. D. and Vecchi, M. P. (1983). Optimization by simulated annealing. Science, 220: 671–680.
Kondrashov, A. S. (1984). On the intensity of selection for reproductive isolation at the beginnings of sympatric speciation. Genetika, 20: 408–415.
Kondrashov, A. S. and Kondrashov, F. A. (1999). Interactions among quantitative traits in the course of sympatric speciation. Nature, 400: 351–354.
Kruuk, L. E. B. (1997). Barriers to Gene Flow: A Bombina (Fire-bellied Toad) Hybrid Zone and Multilocus Cline Theory. Ph.D. Thesis, University of Edinburgh.
Kruuk, L. E. B., Baird, S. J. E., Gale, K. S. and Barton, N. H. (1999). The effect of endogenous and exogenous selection on multilocus clines. Genetics, 153: 1959–1971.
Langley, C. H. (1977). Nonrandom associations between allozymes in natural populations of Drosophila melanogaster. In: Christiansen, F. B. and Fenchel, T. M. (eds) Measuring Selection in Natural Populations, pp. 265–273. Springer Verlag, Berlin.
Li, W. H. and Nei, M. (1974). Stable linkage disequilibrium without epistasis in subdivided populations. Theor Pop Biol, 6: 173–183.
Long, J. C., Williams, R. C. and Urbanek, M. (1995). An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet, 56: 799–810.
Maccallum, C. J., Nurnberger, B., Barton, N. H. and Szymura, J. M. (1998). Habitat preference in the Bombina hybrid zone in Croatia. Evolution, 52: 227–239.
Mangel, M. and Hilborn, R. (1996). The Ecological Detective. Princeton University Press, Princeton, NJ.
Metropolis, N., Rosenbluth, A., Rosenbluth, M., Teller, A. and Teller, E. (1954). Equation of state calculations by fast computing machines. J Chem Phys, 21: 1087–1095.
Phipps, T. E. and Brill, M. H. (1995). Bayesian entropy and inference. Physics Essays, 8: 615–625.
Shpak, M. and Kondrashov, A. S. (1999). Applicability of the hypergeometric phenotypic model to haploid and diploid populations. Evolution, 53: 600–604.
Slatkin, M. and Excoffier, L. (1996). Testing for linkage disequilibrium in genotypic data using the Expectation-Maximization algorithm. Heredity, 76: 377–383.
Sober, E. (1983). Parsimony in systematics: philosophical issues. Ann Rev Ecol Syst, 14: 335–358.
Turelli, M. and Barton, N. H. (1994). Genetic and statistical analyses of strong selection on polygenic traits: what, me normal?. Genetics, 138: 913–941.
Acknowledgements
This work was supported by grant MMI09726 from the BBSRC/EPSRC, and by the Darwin Trust of Edinburgh. I am grateful to W. G. Hill, L. Kruuk and M. Orive, and to the referees, for their helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barton, N. Estimating multilocus linkage disequilibria. Heredity 84, 373–389 (2000). https://doi.org/10.1046/j.1365-2540.2000.00683.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2000.00683.x
Keywords
This article is cited by
-
Contribution of an additive locus to genetic variance when inheritance is multi-factorial with implications on interpretation of GWAS
Theoretical and Applied Genetics (2013)
-
Population genetic structure of Picea engelmannii, P. glauca and their previously unrecognized hybrids in the central Rocky Mountains
Tree Genetics & Genomes (2013)
-
Identification of recent hybridization between gray wolves and domesticated dogs by SNP genotyping
Mammalian Genome (2013)
-
Widespread introgression does not leak into allotopy in a broad sympatric zone
Heredity (2011)
-
Preliminary analysis of a hybrid zone between two subspecies of Zootermopsis nevadensis
Insectes Sociaux (2009)
