
Abstract
Erdheim-Chester disease (ECD) is a rare, distinct clinicopathologic entity with nearly pathognomonic radiographic features. The lesions consist of lipid-storing CD68 (+), CD1a (−) non-Langerhans' cell histiocytes, either localized to the bone or involving multiple organ systems in the body. Whether these histiocytic proliferations represent monoclonal neoplastic populations or are part of a polyclonal reactive process is unclear. We present a case report of ECD in a 35-year-old African-American woman with a progressive course over 6 years. We investigated the clonality of the histiocytes using the HUMARA assay on paraffin-embedded tissue sections but did not find any evidence that these cells represent a monoclonal population. In this report, the characteristics of ECD are reviewed, the genetic basis of the HUMARA assay is discussed, and our results in the context of other clonality investigations reported in the literature to date are summarized.
Similar content being viewed by others
INTRODUCTION
Erdheim-Chester disease (ECD) is a rare disease with distinct clinicopathologic and radiographic features and unknown etiology. In 1930, William Chester and his mentor, the Viennese pathologist Erdheim, first described two patients with a distinctive form of lipoid granulomatosis that they regarded as different from other histiocytic disorders, particularly Hand-Schuller-Christian and Niemann-Pick diseases (1). In 1972, Jaffe reported a similar case and named this disease after Erdheim and Chester (2).
ECD is caused by xanthogranulomatous infiltration of numerous foamy non-Langerhans' cell, lipid-laden histiocytes. It has nearly diagnostic radiological changes characterized by bilateral osteosclerosis that involves the long bone metaphyses and diaphyses symmetrically but spares the epiphyses. The most common sites involved are the distal femur and proximal tibia and fibula. The clinical spectrum ranges from focal bone lesions to multisystemic, life-threatening involvement of the visceral organs.
There is an ongoing debate regarding the origin of the non-Langerhans' cell histiocytes in ECD (3, 4, 5, 6). The clinicopathological and radiological features of this disease are different from those of Langerhans' cell histiocytosis (LCH; 7, 8, 9, 10, 11), which is characterized by the proliferation of S100+ and CD1a+ Langerhans' cells. However, several cases of patients with both conditions (12, 13), as well as a case of ECD with eosinophilic granuloma (14) have been reported in the literature, suggesting that there may be a link between these entities. All forms of Langerhans' cell histiocytosis were recently proven to be monoclonal using different X-linked polymorphic DNA probes, including the HUMARA assay, in lesional tissues (15, 16). Few attempts had been made to determine the clonality of the non-Langerhans' cell histiocytes in ECD (17). The aim of the authors was to determine the clonality of these cells from both the bone marrow and pericardial biopsies in a 35-year-old woman with ECD, using the HUMARA assay.
CASE HISTORY
A 35-year-old African-American woman was referred to Shands Hospital at the University of Florida (STH), in 1995 with a complaint of intermittent bone pain. Radiographic skeletal survey showed diffuse bilateral osteosclerosis involving the appendicular skeleton. It was most pronounced about the knees (Fig. 1A), ankles, elbows, distal radii, and ulnae bilaterally but spared the thoracic spine. A bone scan showed bilateral symmetrical increased uptake in the femoral and tibial condyles, as well as about the ankles. Abnormal symmetrical uptake was also seen about the shoulders, elbows, and distal radii and ulnae bilaterally. Magnetic resonance imaging of both knees showed diffuse marrow replacement in the visualized femurs, tibias and fibulas, with incomplete, spotty sparing of the subchondral bone of the epiphysis (Fig. 1B). Histologic sections from a distal left femur core biopsy, done at the outside hospital, showed osteonecrosis with intertrabecular fibrosis. There was also a suggestion of histiocytes within the intertrabecular tissue. She underwent an incisional biopsy of the right proximal tibia in 1996. Both the radiological and light microscopic features of the bone marrow biopsy specimen were consistent with ECD disease. She was treated with three cycles of vincristine. Her clinical course was complicated by chronic pancreatitis with pancreatic pseudocyst, bile duct stricture, and hepatitis C. She had a laparoscopic cholecystectomy and choledochoduodenostomy in 1996. Recurrent pericardial effusion and cardiac tamponade developed, requiring a temporary pericardial drain placement in 1998.

A, plain radiograph of the distal right femur demonstrates irregular areas of radiodensity involving the medullary cavity (arrows) as well as slight cortical thickening. B, coronal magnetic resonance MR image (TE = 15, TR = 483, fat suppression) demonstrates diffuse marrow replacement in the femora and tibiae, with multifocal changes suggestive of infarcts.
She was readmitted to Shands Hospital in 1999 for evaluation of abnormal liver enzymes and was found to have ascites, hepatomegaly, and bilateral pitting edema. A few days later, she developed a large pericardial effusion with cardiac tamponade and hemodynamic compromise. A pericardial window was created, at which time pericardial fluid was drained and pericardial biopsy was obtained. The biopsy, similar to the previous tibial biopsy in 1996, demonstrated changes consistent with ECD. A liver biopsy was obtained and showed no evidence of ECD. Treatment was started with prednisone and vincristine for 7 months, after which the patient decided to stop therapy. During that period, the patient seemed to have worsening symptoms. Chemotherapy was restarted with prednisone and vinblastine. Treatment was complicated by steroid-induced hyperglycemia. Thus far, the patient has had no other complications.
MATERIALS AND METHODS
Tissue Samples
Sections from archival formalin-fixed, decalcified, and paraffin-embedded bone marrow biopsy stained with hematoxylin and eosin (H&E) and toluidine blue were reviewed. Tissue obtained from the pericardial biopsy specimens was fixed in 10% buffered formalin and routinely processed, sectioned and stained with H&E and acid-fast stain. A frozen section was also stained with Oil-Red-O at the time of surgery.
Immunohistochemistry
Paraffin-embedded, formalin-fixed 4 μm sections from pericardial tissue were mounted on positively charged glass slides. They were immunostained according to established protocols using 3,3′-diaminobenzidine detection kit (indirect biotin-avidin system) and an automated immunostainer (Ventana Medical Systems, Tucson, AZ). Immunoreactive epitope retrieval was performed by the heat induced epitope retrieval procedure with Trilogy (Cell Marque Corporation, Hot Springs, AR). We used antibodies against S100 and CD68, diluted 1:1000 (DAKO Corporation, Carpinteria, CA) and CD1a, diluted 1:2 (Immunotech, Westbrook, ME). Internal and external controls and irrelevant antibodies in each staining run were acceptable.
Ultrastructural Examination
Representative portions from the formalin-fixed and paraffin-embedded lesional pericardial tissues were selectively sent for electron-microscopic examination.
Analysis of Clonality:
To determine whether the ECD was a polyclonal reactive process or a monoclonal neoplastic disorder, we studied the clonal status of the non-Langerhans' cell lipid-laden histiocytes using a polymerase chain reaction (PCR) based DNA analysis of the patterns of X-chromosome inactivation at the human androgen receptor gene (HUMARA assay). To accomplish this, DNA was extracted from formalin-fixed, paraffin-embedded unstained 4–5 μm lesional tissue sections. To reduce the effect of contaminating inflammatory and stromal cells, the lesional pericardial areas were dissected from the unstained tissue section using a 21-gauge needle. The histiocytes in the tissue submitted comprised >50% of the cells by microscopic estimate. DNA from the uninvolved liver biopsy of the patient was used as normal control tissue.
Analysis of the inactivation pattern of the HUMARA locus was performed using PCR by one of the authors (JB) at the Biomolecular Laboratory, Children's Mercy Hospital, Kansas City, Missouri, according to their standard protocol using the primers of Allen et al. (18, 19). In this procedure, DNA was digested initially with one of two methylation-sensitive restriction enzymes. This prestep ensured that in the ensuing PCR reaction, a product would be obtained only when the X chromosome was inactive. Detection of the paternal versus maternal alleles then relies on the heterozygosity associated with the polymorphic CAG repeat. Briefly, DNA was isolated from the histologic sections of unstained tissues using an extended proteinase K incubation, then digested with 20 U of HpaII and DdeI. An aliquot of 2 μL was then added to 1 μm of primers, 250 μm of dNTPs, 0.05 U of Taq polymerase/μL, and 2.5 mm of MgCl2. PCR included an initial denaturation at 95° C for 1 minute, followed by cycles of 96° C for 20 seconds; 60° C for 30 seconds; 72° C for 1 minute, repeated 34 times; followed by extension at 72° C for 2 minutes and a 10° C hold. The products were analyzed using 10% PAGE (polyacrylamide gel electrophoresis) with pGEM molecular weight standards and SyBr-gold colorimetric detection.
RESULTS
Histologic Findings
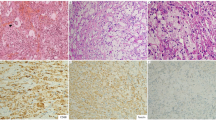
The right proximal tibial bone marrow biopsy, taken in 1996, demonstrated evidence of previous bony destruction and repair. There was complete replacement of the fatty marrow by a variable degree of fibrous tissue and prominent proliferation of foamy histiocytes, including multinucleated forms. Scattered Touton giant cells were present (Fig. 2A). The histiocytes had abundant pale staining and foamy or finely granular cytoplasm without well-formed granulomas (Fig. 2B). Toluidine blue stain for mast cells was negative. The pericardium, from the current material, was thickened (>1 cm) with fibrosis and chronic mononuclear inflammatory cell infiltrate, including a combination of histiocytes, lymphocytes, and plasma cells, as well as scattered multinucleated giant cells. The histiocytes had abundant pale staining and foamy cytoplasm (Fig. 2C). An Oil-red-O stain performed on a frozen section was positive (Fig. 2D). Acid-fast stain was negative for acid-fast bacilli. The liver biopsy revealed no evidence of ECD or granulomatous disease.

A, medium magnification photomicrograph of bone biopsy (200 ×) showing complete replacement of the fatty marrow by a variable degree of fibrous tissue and prominent proliferation of foamy histiocytes including multinucleated forms. Scattered Touton giant cells (inset) are present. B, the histiocytes in the bone biopsy have abundant pale staining and foamy or finely granular cytoplasm without well-formed granulomas (400 ×). C, low-magnification photomicrograph of the pericardial biopsy (100 ×) showing fibrosis and prominent histiocytic infiltrate with mononuclear inflammatory cells. The histiocytes (inset) have abundant pale staining and foamy or finely granular cytoplasm. D, oil-red-O stain performed on a frozen section of pericardium is positive (600 ×). E–G, high-magnification photomicrographs of pericardium (400 ×) illustrating almost the same field on serial sections, immunostained for CD68 (E), CD1a (F), and S-100 (G).
Immunohistochemical stains
Sections from the pericardium showed that the foamy histiocytes were CD1a (−), CD68 (+), and partially S100 positive (Fig. 2E–G).
Electron Microscopy
Ultrastructural examination revealed histiocytes and multinucleated giant cells lacking Birbeck granules. They had clear cytoplasmic spaces that most likely represented the lipids lost during conventional processing. Possible lysosomes were noted in the cytoplasm.
Clonality Results
The control tissue (liver) yielded an informative PCR pattern demonstrating heterozygosity at the HUMARA site in this patient. The lesional tissue from the pericardium demonstrated a similar PCR product pattern in comparison to the control liver tissue. Because a skewed inactivation pattern was not detected in the pericardial tissue, these results were interpreted as indicating a lack of monoclonality.
DISCUSSION
The clinical presentation, radiographic findings, bone marrow and pericardial morphology, immunohistochemical profile, and ultrastructural features in the case presented are all consistent with ECD. The patient presented with bone pain and had the pathognomonic radiographic features of the disease, which was confirmed by the bone biopsy. She also had evidence of extraskeletal manifestations that was proven by the pericardial biopsy.
ECD usually affects men and women equally in a wide age range, mostly the middle aged or elderly. It is usually diagnosed on the basis of the nearly pathognomonic radiographic features showing bilateral symmetrical osteosclerosis of the long bones, almost always sparing the epiphyses. However, epiphyseal involvement has been previously reported (20). Approximately one third of the patients have additional lytic bone changes. Hand-Schuller-Christian disease, in contrast, is characterized by bony involvement with Langerhans' cells that produce lytic lesions. Membranous bones (skull, ribs, and pelvis) are usually affected. Extraskeletal manifestations of ECD include hypothalamic/posterior pituitary dysfunction such as diabetes insipidus and involvement of the retroperitoneum and pericardium (21, 22). ECD can lead to heart failure due to myocardial or pericardial histiocytic infiltration, as seen in this case. The cerebral manifestations are variable and clinically are similar to multiple sclerosis. Lesions can be located in the cerebral hemispheres, hypothalamus, cerebellum, brain stem, and meninges (21). Lesions in the choroid plexus have also been described. Orbital disease with exophthalmos and xanthoma-like lesions of the eyelid are also common (5). Pulmonary involvement is rare, but advanced lesions are associated with extensive fibrosis that can lead to cardiorespiratory failure (9, 11, 23). No effective treatment strategies have been described in the literature thus far. The prognosis depends on the extent and distribution of the extra-skeletal disease. Veyssier-Belot et al. reviewed 59 cases and concluded that 22 (59%) of 37 patients with follow-up died of the disease, 8 within a 6-month period (23).
The immunohistochemical profile of the lesional cells is histiocytic (CD68+). In the case described, there was some variable S-100 protein reactivity, but CD1a was negative. The latter is an important feature in differentiating ECD from LCH. Langerhans' cell histiocytosis is characterized by the proliferation of monoclonal CD1a+ and S100+ Langerhans' cells (15, 16). The absence of CD1a immunoreactivity and Birbeck granules help distinguish our case of ECD from LCH. To our best knowledge, CD1a was uniformly negative in all cases of ECD reported to date. In ECD, the lesional cells are usually S-100 negative, although positivity has been described in previous cases in the literature and in different organs in patients with otherwise typical disease (9, 11, 20). The histiocytes in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) are similar to those seen in ECD, but they frequently contain intact lymphocytes (emperipolesis). Moreover, they are consistently S-100 (+) and CD1a (−). Storage diseases such as Gaucher's disease are easily differentiated from ECD on the basis of associated clinical findings. Morphologically, Gaucher cells have a distinctive appearance that is different from that of the finely vacuolated histiocytes in ECD. The former are distended by pale fibrillary cytoplasmic inclusions resulting from lysosomal accumulation of glucocerebrosides.
Monoclonality is one of the hallmarks of neoplasia. In contrast, reactive processes are polyclonal. X-chromosome inactivation patterns have been used to determine monoclonality in various tumors. During embryogenesis in females, each somatic cell randomly methylates and thereby inactivates either the maternally derived or the paternally derived X chromosome. The active X-chromosome may be distinguished from the inactive X-chromosome by its state of methylation or by gene expression. This pattern of permanent inactivation (Lyonization) is transmitted to all progeny cells so that normal tissues in females are composed of cellular mosaics, differing in which one of the X-chromosomes is inactivated (24). In contrast, neoplasms in females show a uniform pattern with exclusive inactivation of one X-chromosome because all cells are descendants from a single cell (monoclonal). The HUMARA assay has been recently extensively used by other investigators to determine the clonality of many tumors including Langerhans” cell histiocytosis (15, 16), desmoid fibromatosis (25), dermatofibroma (26), Kaposi sarcoma (27), and renal sporadic angiomyolipoma (28), which all have been proven to be monoclonal diseases. Analysis of the highly polymorphic short tandem repeats (CAG trinucleotide repeats) in exon 1 of the human androgen receptor gene (HUMARA assay) has proven to be much more useful and informative than previously used methods. Its high frequency of polymorphism (allelic variability) and 90% heterozygosity are advantageous features. The PCR products are of different sizes and can be separated with electrophoresis. The methylation status is discriminated with either methylation-sensitive restriction enzymes or sodium bisulfate modification (18, 19). Assessment for clonality is feasible in a high percentage of both formalin-fixed and frozen archival material with this method (18, 30). Other methods for demonstrating monoclonality such as G6PD isoenzymes require fresh tissue, and the X-linked G6PD gene has low frequency of heterozygosity. The value of analysis of the methylation patterns of the X-chromosome–linked phosphoglycerate kinase (PGK) gene in females is also limited by the low frequency of polymorphisms (15, 18, 29, 30).
X-linked inactivation approaches are limited by dilution of tumor cells with polyclonal non-tumor stromal and inflammatory cells. It has been reported that the HUMARA assay can assess clonality only if the monoclonal cells comprise >20% of the total cell population in the specimen submitted. If the inflammatory and the normal stromal cells are >30% of the total cell population, selective extraction of DNA from lesional areas in the formalin-fixed and paraffin-embedded tissue sections should be done to enrich the sample with tumor cells (21). In our case we selectively dissected the areas highly infiltrated by the non-Langerhans' histiocytes. The use of a highly selective microdissection technique such as laser capture microdissection (LCM) to collect cells from stained or unstained formalin-fixed and paraffin-embedded sections may help to minimize the dilutional effect of the polyclonal non-tumor stromal and inflammatory cells. This is an approach we are considering for further study of our case. Feist et al. utilized LCM in conjunction with the HUMARA assay to analyze cells from intraductal mammary carcinoma. The laser beam destroyed the DNA in surrounding stromal cells. To avoid preferential amplification of exclusively one X-chromosome allele in small samples, a certain number of cells and DNA is required (31).
Monoclonal X-chromosome inactivation is not always indicative of monoclonal origin of a tumor, i.e., neoplasia (32). Predominance of a single clone of cells, from a polyclonal or multicellular origin can give rise to a monoclonal pattern. Moreover, a secondary clone forming a minor component of a tumor can simulate the presence of normal benign tissue.
In our case, both the lesional pericardial tissue and the normal control liver tissue showed no evidence of monoclonality, the histiocytes represented >50% of the total cell population in the tissue samples submitted, and the normal control liver tissue had a heterozygous pattern. For these reasons, we assert that the results of the analysis are valid. The clinical history of the patient spans 5 years, and thus far she is doing relatively well. This may support the reactive nature of the lesions. Chetritt et al. (17) studied the clonality of the non-Langerhans' cell histiocytes in ECD using the HUMARA assay. Their results showed that the histiocytes in three patients were monoclonal, one was polyclonal, and one was homozygous for the gene and therefore was not informative.
In summary, ECD is a rare distinct clinicopathologic entity. It has nearly pathognomonic radiographic features and is comprised of lipid-storing CD68 (+), S100 (variable), CD1a (−) histiocytes. Although it has some clinical features similar to Langerhans' cell histiocytosis, the radiographic findings, morphologic features, immunohistochemical profile, and ultrastructural features confirm that it is not a Langerhans' cell or antigen-presenting cell process. Whether ECD is a polyclonal reactive process or a monoclonal neoplastic disorder cannot be yet determined by the limited available data. The cause and pathogenesis of this disorder remain unclear, and further studies are warranted.
References
Chester W . Uber lipidgranulomatose. Virchows Arch A Pathol Anat Histol 1930; 279: 561–602.
Jaffe HL . Gaucher's disease and certain other inborn metabolic disorders: lipid (cholesterol) granulomatosis. In: Jaffe HL, ed. Metabolic and inflammatory disease of the bone and joints. Philadelphia, PA: Lea & Febiger; 1972.p. 535–541.
Brower AC, Worsham GF, Dudley AH . Erdheim-Chester disease: a distinct lipoidosis or part of the spectrum of histiocytosis? Radiology 1984; 151: 35–38.
Evans S, Williams F . Case report. Erdheim-Chester disease: polyostotic sclerosing histiocytosis. Clin Radiol 1986; 37: 93–96.
Van der Lee I, Slee PH, Elbers JR . A patient with diabetes insipidus and periorbital swellings; Erdheim-Chester disease. Neth J Med 1999; 55: 76–79.
Devouasoux G, Lantuejoul S, Chatelain P, Brambilla E, Brambilla C . Erdheim-Chester disease: a primary macrophage cell disorder. Am J Respir Crit Care Med 1998; 157: 650–653.
Resnick D, Greenway G, Genant H, Brower K, Haghighi P, Emmett M . Erdheim-Chester disease. Radiology 1982; 142: 289–295.
Fink MG, Levinson DJ, Brown NL, Sreekanth S, Sobel GW . Erdheim-Chester disease. Case report with autopsy findings. Arch Pathol Lab Med 1991; 115: 619–623.
Egan AJ, Boardman LA, Tazelaar HD, Swensen SJ, Jett JR, Yousem SA, et al. Erdheim-Chester disease: clinical, radiologic, and histopathologic findings in five patients with interstitial lung disease. Am J Surg Pathol 1999; 23: 17–26.
Kenn W, Eck M, Allolio B, Jakob F, Illg A, Marx A, et al. Erdheim-Chester disease: evidence for a disease entity different from Langerhans' cell histiocytosis? Three cases with detailed radiological and immunohistochemical analysis. Hum Pathol 2000; 31: 734–739.
Rush WL, Andriko JA, Galateau-Salle F, Brambilla E, Brambilla C, Ziany-bey I, et al. Pulmonary pathology of Erdheim-Chester disease. Mod Pathol 2000; 13: 747–754.
Boralevi F, Leaute-Labreze C, Tison F, Bioulac-Sage P, Vital C, Delbrel X, et al. Langerhans'-cell histiocytosis and Erdheim-Chester disease: probably not a fortuitous association. Ann Dermatol Venereol 1998; 125(5): 335–338.
Vital C, Bioulac-Sage P, Tison F, Rivel J, Begueret H, Gomez C, et al. Brain stem infiltration by mixed Langerhans' cell histiocytosis and Chester-Erdheim disease: more than just an isolated case? Clin Exp Pathol 1999; 47: 71–76.
Strouse PJ, Ellis BI, Shifrin LZ, Shah AR . Case report 710: symmetrical eosinophilic granuloma of the lower extremities (proven) and Erdheim-Chester disease (probable). Skeletal Radiol 1992; 21: 64–67.
Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans”-cell histiocytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med 1994; 331: 154–160.
Yu RC, Chu C, Buluwela L, Chu AC . Clonal proliferation of Langerhans' cells in Langerhans' cell histiocytosis. Lancet 1994; 343: 767–768.
Chetritt J, Paradis V, Dargere D, Adle-Biassette H, Maurage CA, Mussini JM, et al. Chester-Erdheim disease: a neoplastic disorder. Hum Pathol 1999; 30: 1093–1096.
Allen RC, Zoghbi HY, Moseley AB, et al. Methylation of HpaII and HpaI near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–1239.
Frank TS, Svoboda-Newman SM, Hsi ED . Comparison of methods for extracting DNA from formalin-fixed paraffin sections for nonisotopic PCR. Diagn Mol Pathol 1996; 5: 220–224.
Athanasou NA, Barbatis C . Erdheim-Chester disease with epiphyseal and systemic disease. J Clin Pathol 1993; 46: 481–482.
Wright RA, Hermann RC, Parisi JE . Neurological manifestations of Erdheim-Chester disease. J Neurol Neurosurg Psychiatry 1999; 66: 72–75.
Trito NA, Weinrib S, Kaye TB . Endocrine manifestations of Erdheim-Chester disease (a distinct form of histiocytosis). J Intern Med 1998; 244: 529–535.
Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease: clinical and radiologic characteristics of 59 cases. Medicine 1996; 75: 157–169.
Gartler SM, Riggs AD . Mammalian X-chromosome inactivation. Annu Rev Genet 1983; 17: 155–183.
Lucas DR, Shroyer KR, McCarthy PJ, Markham NE, Fujita M, Enomoto TE . Desmoid tumor is a clonal cellular proliferation: PCR amplification of HUMARA for analysis of patterns of X-chromosome inactivation. Am J Surg Pathol 1997; 21: 306–311.
Chen TC, Kuo T, Chan HL . Dermatofibroma is a clonal proliferative disease. J Cutan Pathol 2000; 27(1): 36–39.
Delabesse E, Oksenhendler E, Lebbe C, et al. Molecular analysis of clonality in Kaposi's sarcoma. J Clin Pathol 1997; 50: 664–668.
Paradis V, Laurendeau I, Vieillefond A, et al. Clonal analysis of renal sporadic angiomyolipomas. Hum Pathol 1998; 29: 1063–1067.
Mashal RD, Lester SC, Sklar J . Clonal analysis by study of X chromosome inactivation in formalin-fixed paraffin-embedded tissue. Cancer Res 1993; 53: 4676.
Enomoto T, Fujita M, Inoue M, et al. Analysis of clonality by amplification of short tandem repeats. Diagn Mol Pathol 1994; 3: 292–297.
Feist H, Lilischkis R, Hallas C, et al. Clonal analysis in cells using PCR and laser microdissection. Verh Dtsch Ges Pathol 1997; 81: 339–342.
Fialkow PJ . Clonal origin of human tumors. Biochim Biophys Acta 1976; 458: 283–321.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Al-Quran, S., Reith, J., Bradley, J. et al. Erdheim-Chester Disease: Case Report, PCR-Based Analysis of Clonality, and Review of Literature. Mod Pathol 15, 666–672 (2002). https://doi.org/10.1038/modpathol.3880583
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880583
Keywords
This article is cited by
-
Role of multimodality imaging in the diagnosis of lymphoproliferative malignancies and hematologic disorders of the kidneys
Abdominal Radiology (2017)
-
Erdheim-Chester- und Rosai-Dorfman-Erkrankung
Der Pathologe (2015)
-
Spectrum of abdominal imaging findings in histiocytic disorders
Abdominal Imaging (2015)
-
Clinical considerations and key issues in the management of patients with Erdheim-Chester Disease: a seven case series
BMC Medicine (2014)
-
Erdheim–Chester disease: An unusual presentation of an uncommon disease
Skeletal Radiology (2014)
