
Abstract
Two independent pathways of vulvar carcinogenesis have currently been identified, one related to infection with mucosal human papillomaviruses (HPVs) and a second related to chronic inflammatory or autoimmune processes. The goal of the study was to examine a possible role of cutaneous HPVs from the beta genus in vulvar carcinogenesis and to evaluate the distribution of intratypic variants of HPV 16 in HPV 16-positive vulvar cancer. Consecutive cases of vulvar carcinoma were retrieved from the files and included the following histologic subtypes: keratinizing (n=21), basaloid (n=7), warty (n=1), mixed basaloid–warty (n=4), verrucous (n=4), keratoacanthoma (n=1), basal cell carcinoma (n=1). All tumors were microdissected and tested for 25 beta HPV types and 25 mucosal HPV types. Cases identified as positive for HPV 16 were further tested for intratypic variants. All cases were immunostained for p16INK4a. Beta HPVs were not detected in any of the tumor cases. Mucosal HPVs were detected in all but one basaloid/warty carcinomas; of these, nine cases (82%) were positive for HPV 16, including five European subtypes, one African subtype, one North American subtype and two indeterminate subtypes. Two of four verrucous carcinomas were positive for HPV 6. Mucosal HPVs were not detected in keratinizing carcinomas, keratoacanthoma and basal cell carcinoma. All cases of basaloid/warty carcinomas, but none of the remaining tumors, overexpressed p16INK4a protein. Our data do not support a role of beta HPVs in the pathogenesis of vulvar carcinoma. The study reaffirms the role of mucosal HPVs, in particular that of HPV 16, in the pathogenesis of basaloid and warty tumor subtypes. The HPV 16 intratypic variation showed correlation with patients’ ethnic background. P16INK4a immunostaining seems to be a sensitive and specific marker of vulvar carcinomas positive for oncogenic mucosal HPVs.
Similar content being viewed by others

Main
The pathogenesis of vulvar carcinoma is not entirely understood. Results of epidemiologic, clinicopathologic and virologic studies are in support of two independent pathways of vulvar carcinogenesis, one related to infection with oncogenic mucosal human papillomaviruses (HPVs) and a second related to a chronic inflammatory and/or autoimmune process involving vulvar mucosa and skin.1, 2, 3, 4, 5, 6
Vulvar cancers associated with oncogenic mucosal HPVs tend to occur in young women with past history of genital warts, cervical dysplasia and/or immunosuppression. These carcinomas typically belong to basaloid or warty histologic subtypes and develop from a well-characterized in situ lesion, namely, classic vulvar intraepithelial neoplasia grade 3 (VIN3).7 Approximately 80–90% of HPV-positive vulvar cancers are associated with HPV16 and nearly all remaining tumors are positive for HPV18 and HPV33.4, 5, 6, 8, 9, 10, 11, 12, 13
Vulvar carcinomas not associated with mucosal HPVs occur in older women. These well-differentiated keratinizing squamous cell carcinomas histologically resemble conventional squamous cell carcinomas occurring in non-genital skin.4 However, unlike the usual squamous cell carcinomas, vulvar cancers are not arising in the setting of actinic keratosis. In the vulva, these tumors develop in a background of squamous cell hyperplasia, a chronic inflammatory condition, or long-lasting lichen sclerosus, a lesion with strong links to autoimmune diseases. Although patients with lichen sclerosus have an average 4.5% cumulative risk of vulvar squamous cell carcinomas (reviewed in Carlson 1998, Table 6),14 neither lichen sclerosus nor squamous cell hyperplasia are universally accepted to be premalignant lesions.1, 14, 15 Differentiated VIN or VIN simplex is recognized as an immediate precursor of invasive keratinizing squamous cell carcinomas in the vulva.1, 16, 17 Since differentiated VIN is almost invariably seen in association with squamous cell hyperplasia and lichen sclerosus, it is thought to represent a histologically distinct phase of malignant transformation in a non-neoplastic epithelial disorder, such as squamous cell hyperplasia or lichen sclerosus.17, 18 The consecutive molecular events of malignant transformation in lichen sclerosus, squamous cell hyperplasia and differentiated VIN leading to invasive squamous cell carcinomas are currently unknown. A report of allelic imbalance in squamous cell hyperplasia and lichen sclerosus supports the hypothesis that both conditions pose a risk for neoplasia despite the lack of morphologic evidence of atypia.18 Rare cases of lichen sclerosus and adjacent squamous cell carcinomas with identical p53 gene mutations have been reported.19 Overall, however, p53 gene mutation is an infrequent and rather late event in vulvar carcinogenesis.11, 15, 20
As reviewed by Nindl et al,21 there is evidence supporting a role for cutaneous HPV types belonging to the beta-genus (betaPVs) (Table 1) in the pathogenesis of non-melanoma skin cancer. Originally, betaPVs have been found in benign and malignant skin lesions from patients with a rare hereditary disease, epidermodysplasia verruciformis. These individuals develop multifocal skin warts and maculae that have a high rate of progression to squamous cell carcinomas, in particular on sun-exposed sites. Specific betaPVs, that is HPV5 and HPV8, have been suggested as possible high risk types in these patients.22 Subsequently, studies of betaPV types showed their presence in squamous cell carcinomas of immunosuppressed individuals23 and in skin cancers of immunocompetent individuals.23, 24, 25 Cutaneous HPVs, however, are ubiquitous and are persistently present on the surface of normal skin and in plucked hairs.26, 27, 28, 29, 30 Whether the detection of betaPVs in squamous cell carcinomas reflects their oncogenic potential or whether it represents merely a persistent cutaneous infection that occurs in a large percentage of the population28 is still unclear.
The groups of vulvar squamous cell carcinomas that may be associated with cutaneous HPVs are keratinizing and verrucous carcinomas. These tumors usually develop in a background of squamous cell hyperplasia and lichen sclerosus, conditions treated with topical corticosteroids producing local immunosuppression. Some of the keratinizing squamous cell carcinomas show areas of cytologic atypia with high resemblance to koilocytic change.13 Furthermore, the characteristic feature of verrucous carcinoma is a prominent exophytic growth resembling a skin wart. It is not inconceivable that this pattern of growth may reflect a proliferation stimulated by cutaneous HPVs. While testing for cutaneous HPVs in keratinizing and verrucous vulvar squamous cell carcinomas has not been reported previously, these tumor subtypes have been previously tested for mucosal HPVs showing either absence or low prevalence of HPV DNA.4, 5, 6, 12, 13 Some studies report the presence of HPV6 and HPV11 in rare cases of verrucous carcinoma.31, 32 Since keratinizing and verrucous vulvar squamous cell carcinomas have never been evaluated for the presence of cutaneous HPVs, the first goal of our study was to test these tumors for the presence of cutaneous HPV types from the beta genus33 and for mucosal HPVs using the SPF10–LiPA25 system detecting 25 low and high oncogenic risk mucosal HPVs.34, 35
Recently, upregulation of p16INK4a, a tumor suppressor protein, has been identified as a sensitive marker of active E7 oncoprotein expression in in situ and invasive carcinomas caused by oncogenic mucosal HPVs.36, 37, 38 P16INK4a immunostaining of such lesions allows for a visual verification of the extent of tissue involved by oncogenic mucosal HPVs. To validate the results of HPV DNA detection by PCR and to exclude the cases with detection of skin surface ‘HPV passengers’, all cases in our study were immunostained for p16INK4a protein.
Oncogenic properties of HPVs mainly depend on two viral oncoproteins, E6 and E7, which interfere with the functions of the host cell p53 tumor suppressor protein and the retinoblastoma protein, respectively. Intratypic variations of HPV16 E6 and E7 genes have been described in which the gene sequence variation results in oncoprotein variants with a transforming potential, which differs from that of the prototype (reviewed by Giannoudis et al39 and Bernard et al40). The second goal of this study was to examine the intratypic variation of HPV16 in vulvar carcinomas.
Materials and methods
Case Selection and Review
Institutional review board approval was obtained for this study. Consecutive cases of vulvar squamous cell carcinomas diagnosed between December 1990 and December 2005 were retrieved from the files of the Pathology Department at the Weill Medical College of Cornell University New York, NY, USA. A total of 39 cases were identified, including keratinizing squamous cell carcinoma (n=21), verrucous (n=4), basaloid (n=7), warty (n=1), mixed basaloid–warty (n=4), keratoacanthoma (n=1) and basal cell carcinoma (n=1). The cases were re-reviewed to confirm the diagnosis and subclassification into the histologic subtypes. The standard diagnostic criteria were used.1 Cases of re-excision or recurrences were excluded from the study.
Tissue Dissection and DNA Preparation
Genomic DNA was prepared from two to three 4 μm sections from each case using standard methods. Briefly, the slides were deparaffinized and tissue was scraped with a sterile blade. To check for contamination representative tumor sections were microdissected into three separate samples:
-
1
deep tumor portion, consisting of deep nests with a minimum of 2 mm distance from the tumor surface,
-
2
superficial tumor portion, consisting of 2 mm superficial layer,
-
3
stroma, a negative dissection control.
DNA was released from the samples by incubating them with 100 μl proteinase K solution (1 mg/ml) for 16 h at 56°C. Following heat inactivation at 95°C for 10 min, 10 μl of the supernatant was used for PCR. Appropriate positive and negative controls were incorporated during DNA preparation and subsequent testing to monitor test performance.
BetaPV Genotyping with the PM-PCR RHA Method
BetaPV detection and genotyping was carried out with the PM-PCR reverse hybridization assay (RHA) method (The skin (beta) HPV prototype research assay; Diassay BV, Rijswijk, The Netherlands) as described earlier by de Koning et al.33 The method was designed for the identification of the 25 established betaPV types (ie, HPV genotypes 5, 8, 9, 12, 14, 15, 17, 19, 20, 21, 22, 23, 24, 25, 36, 37, 38, 47, 49, 75, 76, 80, 92, 93 and 96). The PM-PCR, generating a biotinylated amplimer of 117 bp from the E1 region, was carried out with all precautions to avoid contamination as described by the manufacturer. Briefly, PM-PCR was performed in a final reaction volume of 50 μl, containing 10 μl of the isolated DNA, 2.5 mM MgCl2, 1 × GeneAmp PCR Buffer II, 0.2 mM deoxynucleoside triphosphates, 1.5 U AmpliTaq Gold DNA polymerase and 10 μl of the PM primer mix. The PCR was performed by a 9 min preheating step at 94°C, followed by 35 cycles of amplification comprising 30 s at 94°C, 45 s at 52°C and 45 s at 72°C. The PCR is ended by a final elongation step of 5 min at 72°C. As a positive PCR control, a betaPV plasmid clone was included at an amount of approximately 100 times the limit of detection of the assay. Identification of the amplified HPV types was performed by analyzing the amplimers with the RHA.
Mucosal HPV DNA Amplification and Genotyping
Broad-spectrum HPV DNA amplification and mucosal HPV genotyping was performed using the SPF10–LiPA25 system (SPF10 HPV LiPA, version 1; manufactured by Labo Bio-Medical Products, Rijswijk, The Netherlands) as described previously.34, 35 Briefly, SPF10 PCR amplifies a 65-base pair fragment from the L1 region of the HPV genome. The reaction was performed in a total volume of 50 μl containing 10 μl of isolated DNA, 1 × GeneAmp PCR Buffer II, 0.2 mM deoxynucleoside triphosphates, 15 pmol of each of the forward and reverse primers, and 1.5 U of AmpliTaq Gold DNA polymerase. AmpliTaq Gold was activated by incubation at 94°C for 9 min. HPV DNA was amplified in 40 cycles of 30 s at 94°C, 45 s at 52°C and 45 s at 72°C, and a final extension of 5 min at 72°C. The use of biotinylated reverse primers enabled the capture of the reverse strand of the amplimer onto streptavidin-coated microtiter plates. The captured amplimers were denatured by alkaline treatment, and detected by a defined cocktail of digoxigenin-labeled probes. This method is designated the HPV DEIA, which provides an optical density value and is able to detect more than 50 HPV types. Amplimers from positive samples can be used to identify 25 individual HPV genotypes (high-risk HPV: 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, 70, and low-risk HPV: 6, 11, 34, 40, 42–44, 53, 54, 74) simultaneously in the RHA.
HPV16 Variant Testing
Cases identified as positive for HPV16 were further tested for intratypic subtypes and variants by PCR and subsequent analysis of the sequences of the E6/E7 region by reverse hybridization. The following nucleotide positions were assessed according to the HPV16 reference sequence (accession number K02718): 131, 132, 143, 145, 178, 286, 289, 335, 350, 403 and 532. This method allowed for determination of the European subtype with variants Asian, 350G, 350T, and 131G, the African subtype with variants African 1 and African 2 and the North American/Asian-American subtype with variants North American and Asian American. The nomenclature was based on Yamada et al.41
Reverse Hybridization Assay
In the RHA, biotinylated amplimers were analyzed by hybridization to DNA probes that are attached to nitrocellulose strips in parallel lines. BetaPV and mucosal HPV genotyping was carried out separately. Briefly, 10 μl of the biotinylated amplimer was mixed with 10 μl denaturation solution in a plastic trough containing the genotyping strips. For the betaPV genotyping, additionally 10 μl of 3B-buffer was added. The mix was incubated for 5 min at room temperature. Two milliliters of pre-warmed (37°C) hybridization buffer (3 × SSC, 0.1% sodium dodecyl sulfate) was added and incubated at 50±0.5°C for 1 h. All incubations and washing steps were performed automatically in a ProfiBlot™ 48 T (Tecan Austria GmbH, Salzburg, Austria). The strips were washed twice for 30 s and once for 30 min at 50°C with 2 ml of hybridization solution. Following this stringent wash, the strips were incubated with 2 ml of alkaline phosphatase–streptavidin conjugate for 30 min at room temperature. Strips were washed twice with 2 ml of rinse solution and once with 2 ml of substrate buffer. Two milliliters of substrate (5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium) was added and incubated for 30 min at room temperature. The reaction was stopped by washing for 3 and 10 min with rinse solution and by a wash with 2 ml of water. The strips were dried and the purple colored bands were visually scored and interpreted by aligning with the standard grid.
P16INK4a Immunohistochemistry
P16INK4a immunohistochemical staining was performed on 4 μm sections of formalin-fixed, paraffin-embedded specimens. The sections were subjected to heat-induced antigen retrieval and incubated in an automated stainer with p16INK4a antibodies obtained from CINtec® Histology Kit (MTM Laboratories, Westborough, MA, USA). The sections were subsequently stained using Bond Polymer Detection System (Vision Biosystem, Norwell, MA, USA) developed with diaminobenzidine chromogen and counterstained with hematoxylin. The staining was performed with respective negative and positive controls. The result of the staining was graded as following: 0, negative or weak nuclear and cytoplasmic ‘blush’; 1, moderate to strong intensity nuclear and cytoplasmic staining with diffuse or patchy distribution. According to the manufacturer's instructions, weak ‘blush’ staining should be interpreted as a negative result.
Results
Clinicopathologic Characteristics of Cases
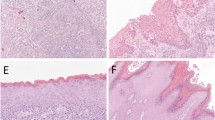
A total of 39 cases of vulvar carcinoma were identified during the study period. Typical keratinizing squamous cell carcinoma accounted for 21/39 (54%) of the cases, whereas combined basaloid, warty and mixed basaloid–warty carcinomas added up to 12/39 (31%) of cases (Table 2). The average age of the patients with basaloid, warty (Figure 1a and b) and mixed basaloid–warty carcinomas was 58 years. The patients with keratinizing and verrucous carcinomas (Figure 1c and d) were on average two decades older (average age 76 years) and the average age difference between the two groups was statistically significant, P=0.0004.

Main histologic subtypes of vulvar carcinoma: (a) basaloid carcinoma, (b) warty carcinoma, (c) keratinizing carcinoma, (d) verrucous carcinoma (hematoxylin and eosin, (a, b) original magnification × 200, (c, d) original magnification × 100).
All cases of basaloid/warty carcinomas were associated with classic VIN3 (Table 2). Sixty-eight percent (17/25) of combined keratinizing and verrucous carcinomas were associated with squamous cell hyperplasia and differentiated VIN; in addition, three of these cases also showed lichen sclerosus. The remaining cases were associated with a single lesion: squamous cell hyperplasia, 3/25 (12%); differentiated VIN, 3/25 (12%) and lichen sclerosus, 2/25 (8%). A single case of keratoacanthoma was associated with squamous cell hyperplasia and differentiated VIN. No non-neoplastic vulvar disorder was identified adjacent to a case of basal cell carcinoma.
Some cases of basaloid carcinomas showed focal morphologic features overlapping with keratinizing carcinoma, and vice versa. For instance, 5 of 12 cases (42%) of basaloid and warty carcinomas showed focal areas of keratin pearl formation (Figure 2a) and 7 of 21 cases (33%) of moderately differentiated keratinizing carcinoma showed areas of tumor nests with scant basophilic cytoplasm and inconspicuous keratinization, resembling basaloid carcinoma (Figure 2b). In addition, 7 of 21 cases of keratinizing squamous cell carcinomas showed areas of cytologic atypia resembling koilocytic change, mimicking warty carcinoma (Figure 2c); in two of such cases adjacent differentiated VIN also showed similar ‘pseudo-koilocytic’ atypia (Figure 2d).

Overlapping features in subtypes of vulvar carcinoma: (a) keratin pearls in basaloid carcinoma, (b) pseudo-basaloid nests in keratinizing carcinoma, (c) pseudo-koilocytic atypia in keratinizing carcinoma, (d) pseudo-koilocytic atypia in differentiated VIN (hematoxylin and eosin, (a–d) original magnification × 200).
The two tumor groups, however, showed distinct differences in nuclear chromatin distribution. Keratinizing/verrucous tumors had relatively pale nuclei with vesicular chromatin and prominent nucleoli, while basaloid/warty tumors showed hyperchromatic nuclei with coarse, stippled chromatin and inconspicuous nucleoli.
Detection of betaPV and Mucosal HPVs
Cutaneous betaPVs were not detected in any of the tumor cases (Table 2). All but one case (case 1) of basaloid, warty and mixed basaloid–warty carcinomas were positive for mucosal HPVs. Microdissection and SPF10–LiPA25 analysis of case 1 was repeated twice and confirmed the original result. Nine cases were positive for HPV16 and two tumors were positive for HPV33 and HPV33 plus HPV51, respectively (Table 2). Analysis of intratypic HPV16 variation revealed five European subtypes, one North American variant and one African1 variant (Table 2). In two cases, the exact subtype/variant could not be determined by the RHA system. The patients with European subtypes were of either Caucasian or Hispanic racial background, the patient with the African1 variant was an Afro-American and the background of the patient with North American variant was not on record. Of two patients with undetermined variants, one was Caucasian and one was Afro-American.
Mucosal HPVs were not detected in any of the keratinizing squamous cell carcinomas, keratoacanthoma or basal cell carcinoma; however, two of four verrucous carcinomas were positive for HPV6.
Comparison of the results of HPV DNA amplification between the three microdissected samples from the same case enabled, by repeat analysis, identification of six cases with amplification contamination: in three cases, tumor surface showed contamination with HPV6 (n=1) and HPV16 (n=2), and three cases showed post-dissection contamination with HPV66.
All PCR assays were controlled by appropriate positive and negative controls. No aberrant results were observed.
P16INK4a Immunostaining
All cases of basaloid, warty and mixed basaloid–warty carcinomas and adjacent VIN3 stained diffusely and strongly with p16INK4a. None of the verrucous, keratinizing squamous cell carcinomas, keratoacanthoma and basal cell carcinoma, adjacent squamous cell hyperplasia, lichen sclerosus or differentiated VIN showed significant p16INK4a positivity with the exception of focal ‘blush’ staining, which was identified in 20% of the cases. Weak ‘blush’ result of p16INK4a immunostaining should be interpreted as a negative result, as explained in the Materials and methods section.
Positive p16INK4a staining showed excellent correlation with the detection of high-risk mucosal HPVs with the exception of one case: case number 1 in Table 2. In this case, the tumor was of basaloid subtype and was associated with classic VIN3 in situ lesion. Based on these histopathologic features, the lesion was expected to be positive for high-risk mucosal HPV; in addition, both the tumor and VIN3 was diffusely and strongly positive for p16INK4a.
Discussion
Cutaneous betaPVs were not detected in any of the vulvar cancer cases in the current study. While this was a first investigation of betaPVs in vulvar carcinoma that will require further corroboration, results of our initial findings suggest that betaPVs are not involved in pathogenesis of vulvar squamous cell carcinomas. The results of this study, however, provide further validation to the hypothesis of two pathogenetic pathways in vulvar carcinogenesis, one of which is related to infection with high-risk mucosal HPVs. In our study all, but one, basaloid and warty carcinomas were found to be positive for mucosal HPVs, with HPV16 alone accounting for 82% of positive cases. The single HPV-negative basaloid carcinoma most likely represented a false-negative error of the PCR assay, based on histopathologic features and presence of p16INK4a overexpression, although the PCR assay tested for more than 50 HPV types.42
The results of studies of HPV16 variants in cervical mucosa demonstrated that non-European HPV16 variants confer a higher risk of prevalent and incident high-grade cervical dysplasia.43 In addition, results of a case-controlled study from Mexico indicated a higher risk of invasive cervical carcinoma conferred by Asian American–North American variants (odds ratio 27.0), as compared to European HPV16 variants (odds ratio 3.4).44 The current study is a first report on intratypic variation of HPV16 in vulvar carcinoma. The HPV16 intratypic variation correlated with patients’ ethnic background and showed a pattern of distribution similar to that reported for cervical squamous cell carcinoma in North America.45 In contrast to previous studies demonstrating increased frequency of non-European HPV16 variants in invasive squamous cell carcinomas of the cervix,44, 46 we did not observe such relationship in our limited sample of HPV16-positive vulvar cancers. Mucosal HPVs were not detected in keratinizing carcinomas, keratoacanthoma or basal cell carcinoma. This result is in agreement with previous studies that used different analytical techniques and demonstrated either no or only low prevalence of mucosal HPVs in these tumors.4, 5, 6, 12, 13 Interestingly, two of four verrucous carcinomas in our study were found to be positive for HPV6. Since HPV6 is a low oncogenic risk HPV type, lesions caused by this HPV are not expected to show overexpression of p16INK4a and, with in situ hybridization assays being insensitive, no independent verification of viral presence in these tumors is available. While in the majority of larger studies of vulvar and penile verrucous carcinomas no HPV DNA has been detected,47, 48, 49 rare cases of verrucous carcinoma positive for low-risk mucosal HPV types have been reported.31, 32, 50 These latter reports faced two criticisms: possible detection of ‘passenger HPVs’ and misclassification of giant condyloma of Buschke–Lowenstein as verrucous carcinoma. In our study, we made the best attempt to dissect deep tumor tissue to avoid contamination from surface HPVs; in addition, a correct diagnosis has been established as no koilocytic atypia has been identified in any of the cases and the tumors were associated with in situ lesions, including squamous cell hyperplasia and differentiated VIN. There is a long-standing controversy with regard to pathogenesis of verrucous carcinoma and its relationship, or lack of thereof, to giant condyloma of Buschke–Lowenstein. Aside from subtle architectural differences, the main distinguishing feature between these two entities is the presence of koilocytic atypia in giant condyloma and absence of koilocytosis in verrucous carcinoma. However, it is also well documented that a significant proportion of genital condylomata shows minimal or no koilocytic atypia at all.51 Given the overlapping histologic features, some authorities regard giant condyloma of Buschke–Lowenstein and verrucous carcinoma as a spectrum of the same process.52 Since condylomata accuminata do not regress spontaneously and, unless excised, will persist for years or even decades, showing decreasing koilocytosis and proliferation while developing increasing superficial keratinization (unpublished observations51), it is not inconceivable that verrucous carcinoma may evolve from a persistent, irritated condyloma. Low oncogenic risk HPVs may initiate the growth process, while subsequent chronic epithelial irritation may contribute to a gradual evolution of malignant phenotype. A strong argument against the connection between giant condylomas and verrucous carcinomas is the fact that patients with giant condylomas are on average 30 years younger than patients with verrucous carcinomas (35–46 years old vs 65–75 years old).49, 53
The current study shows a precise separation between the two main groups of vulvar cancers: basaloid/warty and keratinizing/verrucous carcinomas, and we have found an accurate correlation between the histologic tumor subtype, adjacent in situ lesion, the presence or absence of oncogenic mucosal HPVs and p16INK4a overexpression. Many of the previous studies of vulvar carcinomas showed some degree of non-correlation and overlap between the histopathologic features, HPV detection and p16INK4a over expression.12, 13, 15, 54 The reasons for this apparent lack of correlation in some cases may be multifold: (1) the presence of mixed morphologic features in the different areas of the tumor, resulting in imprecise classification (eg presence of keratin pearls in basaloid carcinoma, presence of pseudo-koilocytes or inconspicuous keratin pearls in keratinizing carcinoma); (2) detection of ‘passenger HPV’ resulting in non-correlating positive result of HPV detection and negative result of p16INK4a staining; (3) failure of HPV DNA amplification resulting in non-correlating negative result of HPV detection and positive result of p16INK4a staining; (4) additionally, a recent study from our laboratory showed that false-positive interpretive error of ‘blush’ p16INK4a immunostaining could result in a lack of correlation with detection of high oncogenic risk HPVs in cervical mucosa.55
The overlapping histologic features in different tumor subtypes were analyzed in detail in a recent study by Santos et al.13 In the series of vulvar carcinomas described by Santos et al, keratin pearls were identified in over a half of basaloid carcinomas and in all warty and keratinizing carcinomas. In addition, ‘koilocytic atypia’ was identified in 43% of HPV-negative keratinizing carcinomas. Cases with mixed histologic features were also identified in our study: keratin pearls were found focally or diffusely in 42% of basaloid/warty carcinomas, while pseudo-koilocytic atypia and pseudo-basaloid nests were found in 33% of keratinizing carcinomas. Both the correlation between the overall tumor morphology and adjacent in situ lesion and nuclear chromatin distribution were very helpful in determining tumor subclassification in the cases with overlapping histologic features. Keratinizing/verrucous tumors exhibited pale vesicular chromatin and prominent nucleoli, while basaloid/warty tumors showed dark, coarse, granular chromatin and inconspicuous nucleoli. Results of our study also show that p16INK4a immunostaining may be used as an objective adjunct test to confirm the diagnosis in challenging cases.
Some pathologists relay on p53 immunostaining to identify differentiated VIN and HPV-negative vulvar cancer. P53 immunoexpression, however, is neither sensitive nor specific marker of differentiated VIN,56 neither it correlates with the actual p53 gene mutation in benign or malignant vulvar squamous epithelium.57, 58 These two problems with p53 immunostaining have been widely illustrated by numerous previous publications11, 15, 19, 20, 56, 57, 58 and in the authors own experience p53 immunostain is not useful in differential diagnosis of HPV-negative vulvar lesions.
The presented data do not support a role for betaPVs in vulvar carcinoma but reaffirm the role of high-risk mucosal HPV types in the pathogenesis of basaloid and warty vulvar carcinoma. Our results corroborate previous findings of Santos et al13 that p16INK4a immunostaining is a sensitive and specific marker of HPV-positive vulvar carcinomas, useful in subclassifying tumors with mixed or overlapping histologic features.
Accession codes
References
Wilkinson EJ . Premalignant and malignant tumors of the vulva. In: Kurman RJ (ed). Blaustein's Pathology of the Female Genital Tract, 5th edn. Springer: New York, 2002, pp 99–122.
Crum CP . Carcinoma of the vulva: epidemiology and pathogenesis. Obstet Gynecol 1992;79:448–454.
Andersen WA, Franquemont DW, Williams J, et al. Vulvar squamous cell carcinoma and papillomaviruses: two separate entities? Am J Obstet Gynecol 1991;165:329–335.
Toki T, Kurman RJ, Park JS, et al. Probable nonpapillomavirus etiology of squamous cell carcinoma of the vulva in older women: a clinicopathologic study using in situ hybridization and polymerase chain reaction. Int J Gynecol Pathol 1991;10:107–125.
Nuovo GJ, Delvenne P, MacConnell P, et al. Correlation of histology and detection of human papillomavirus DNA in vulvar cancers. Gynecol Oncol 1991;43:275–280.
Bloss JD, Liao SY, Wilczynski SP, et al. Clinical and histologic features of vulvar carcinomas analyzed for human papillomavirus status: evidence that squamous cell carcinoma of the vulva has more than one etiology. Hum Pathol 1991;22:711–718.
Kurman RJ, Toki T, Schiffman MH . Basaloid and warty carcinomas of the vulva. Distinctive types of squamous cell carcinoma frequently associated with human papillomaviruses. Am J Surg Pathol 1993;17:133–145.
Hording U, Kringsholm B, Andreasson B, et al. Human papillomavirus in vulvar squamous-cell carcinoma and in normal vulvar tissues: a search for a possible impact of HPV on vulvar cancer prognosis. Int J Cancer 1993;55:394–396.
Iwasawa A, Nieminen P, Lehtinen M, et al. Human papillomavirus in squamous cell carcinoma of the vulva by polymerase chain reaction. Obstet Gynecol 1997;89:81–84.
Kagie MJ, Kenter GG, Zomerdijk-Nooijen Y, et al. Human papillomavirus infection in squamous cell carcinoma of the vulva, in various synchronous epithelial changes and in normal vulvar skin. Gynecol Oncol 1997;67:178–183.
Lee YY, Wilczynski SP, Chumakov A, et al. Carcinoma of the vulva: HPV and p53 mutations. Oncogene 1994;9:1655–1659.
Pinto AP, Signorello LB, Crum CP, et al. Squamous cell carcinoma of the vulva in Brazil: prognostic importance of host and viral variables. Gynecol Oncol 1999;74:61–67.
Santos M, Landolfi S, Olivella A, et al. p16 overexpression identifies HPV-positive vulvar squamous cell carcinomas. Am J Surg Pathol 2006;30:1347–1356.
Carlson JA, Ambros R, Malfetano J, et al. Vulvar lichen sclerosus and squamous cell carcinoma: a cohort, case control, and investigational study with historical perspective; implications for chronic inflammation and sclerosis in the development of neoplasia. Hum Pathol 1998;29:932–948.
Kim YT, Thomas NF, Kessis TD, et al. p53 mutations and clonality in vulvar carcinomas and squamous hyperplasias: evidence suggesting that squamous hyperplasias do not serve as direct precursors of human papillomavirus-negative vulvar carcinomas. Hum Pathol 1996;27:389–395.
Hart WR . Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol 2001;20:16–30.
Yang B, Hart WR . Vulvar intraepithelial neoplasia of the simplex (differentiated) type: a clinicopathologic study including analysis of HPV and p53 expression. Am J Surg Pathol 2000;24:429–441.
Pinto AP, Lin MC, Sheets EE, et al. Allelic imbalance in lichen sclerosus, hyperplasia, and intraepithelial neoplasia of the vulva. Gynecol Oncol 2000;77:171–176.
Vanin K, Scurry J, Thorne H, et al. Overexpression of wild-type p53 in lichen sclerosus adjacent to human papillomavirus-negative vulvar cancer. J Invest Dermatol 2002;119:1027–1033.
Ngan HY, Cheung AN, Liu SS, et al. Abnormal expression or mutation of TP53 and HPV in vulvar cancer. Eur J Cancer 1999;35:481–484.
Nindl I, Gottschling M, Stockfleth E . Human papillomaviruses and non-melanoma skin cancer: Basic virology and clinical manifestations. Dis Markers 2007;23:247–259.
Pfister H . Chapter 8: Human papillomavirus and skin cancer. J Natl Cancer Inst Monogr 2003;31:52–56.
Harwood CA, Surentheran T, McGregor JM, et al. Human papillomavirus infection and non-melanoma skin cancer in immunosuppressed and immunocompetent individuals. J Med Virol 2000;61:289–297.
Stockfleth E, Nindl I, Sterry W, et al. Human papillomaviruses in transplant-associated skin cancers. Dermatol Surg 2004;30:604–609.
Forslund O, Lindelof B, Hradil E, et al. High prevalence of cutaneous human papillomavirus DNA on the top of skin tumors but not in ‘Stripped’ biopsies from the same tumors. J Invest Dermatol 2004;123:388–394.
Antonsson A, Karanfilovska S, Lindqvist PG, et al. General acquisition of human papillomavirus infections of skin occurs in early infancy. J Clin Microbiol 2003;41:2509–2514.
Hazard K, Karlsson A, Andersson K, et al. Cutaneous human papillomaviruses persist on healthy skin. J Invest Dermatol 2007;127:116–119.
de Koning MNC, Struijk L, Bouwes Bavinck JN, et al. Betapapillomaviruses frequently persist in the skin of healthy individuals. J Gen Virol 2007;88:1489–1495.
Kohler A, Forschner T, Meyer T, et al. Multifocal distribution of cutaneous human papillomavirus types in hairs from different skin areas. Br J Dermatol 2007;156:1078–1080.
Boxman IL, Berkhout RJ, Mulder LH, et al. Detection of human papillomavirus DNA in plucked hairs from renal transplant recipients and healthy volunteers. J Invest Dermatol 1997;108:712–715.
Djurdjevic S, Devaja O, Hadzic B . Malignant potential of gigantic condylomatous lesions of the vulva. Eur J Gynaecol Oncol 1999;20:63–66.
Kondi-Paphitis A, Deligeorgi-Politi H, Liapis A, et al. Human papilloma virus in verrucus carcinoma of the vulva: an immunopathological study of three cases. Eur J Gynaecol Oncol 1998;19:319–320.
de Koning M, Quint W, Struijk L, et al. Evaluation of a novel highly sensitive, broad-spectrum PCR-reverse hybridization assay for detection and identification of beta-papillomavirus DNA. J Clin Microbiol 2006;44:1792–1800.
Kleter B, van Doorn LJ, Ter SJ, et al. Novel short-fragment PCR assay for highly sensitive broad-spectrum detection of anogenital human papillomaviruses. Am J Pathol 1998;153:1731–1739.
Kleter B, van Doorn LJ, Schrauwen L, et al. Development and clinical evaluation of a highly sensitive PCR-reverse hybridization line probe assay for detection and identification of anogenital human papillomavirus. J Clin Microbiol 1999;37:2508–2517.
Khleif SN, DeGregori J, Yee CL, et al. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc Natl Acad Sci USA 1996;93:4350–4354.
Serrano M . The tumor suppressor protein p16INK4a. Exp Cell Res 1997;237:7–13.
Riethdorf S, Neffen EF, Cviko A, et al. p16INK4A expression as biomarker for HPV 16-related vulvar neoplasias. Hum Pathol 2004;35:1477–1483.
Giannoudis A, Herrington CS . Human papillomavirus variants and squamous neoplasia of the cervix. J Pathol 2001;193:295–302.
Bernard HU, Calleja-Macias IE, Dunn ST . Genome variation of human papillomavirus types: phylogenetic and medical implications. Int J Cancer 2006;118:1071–1076.
Yamada T, Wheeler CM, Halpern AL, et al. Human papillomavirus type 16 variant lineages in United States populations characterized by nucleotide sequence analysis of the E6, L2, and L1 coding segments. J Virol 1995;69:7743–7753.
van Doorn LJ, Molijn A, Kleter B, et al. Highly effective detection of human papillomavirus 16 and 18 DNA by a testing algorithm combining broad-spectrum and type-specific PCR. J Clin Microbiol 2006;44:3292–3298.
Villa LL, Sichero L, Rahal P, et al. Molecular variants of human papillomavirus types 16 and 18 preferentially associated with cervical neoplasia. J Gen Virol 2000;81:2959–2968.
Berumen J, Ordonez RM, Lazcano E, et al. Asian-American variants of human papillomavirus 16 and risk for cervical cancer: a case–control study. J Natl Cancer Inst 2001;93:1325–1330.
Yamada T, Manos MM, Peto J, et al. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J Virol 1997;71:2463–2472.
Tornesello ML, Duraturo ML, Salatiello I, et al. Analysis of human papillomavirus type-16 variants in Italian women with cervical intraepithelial neoplasia and cervical cancer. J Med Virol 2004;74:117–126.
Nascimento AF, Granter SR, Cviko A, et al. Vulvar acanthosis with altered differentiation: a precursor to verrucous carcinoma? Am J Surg Pathol 2004;28:638–643.
Gualco M, Bonin S, Foglia G, et al. Morphologic and biologic studies on ten cases of verrucous carcinoma of the vulva supporting the theory of a discrete clinico-pathologic entity. Int J Gynecol Cancer 2003;13:317–324.
Masih AS, Stoler MH, Farrow GM, et al. Penile verrucous carcinoma: a clinicopathologic, human papillomavirus typing and flow cytometric analysis. Mod Pathol 1992;5:48–55.
Noel JC, Vandenbossche M, Peny MO, et al. Verrucous carcinoma of the penis: importance of human papillomavirus typing for diagnosis and therapeutic decision. Eur Urol 1992;22:83–85.
Pirog EC, Chen YT, Isacson C . MIB-1 immunostaining is a beneficial adjunct test for accurate diagnosis of vulvar condyloma acuminatum. Am J Surg Pathol 2000;24:1393–1399.
Bogomoletz WV, Potet F, Molas G . Condylomata acuminata, giant condyloma acuminatum (Buschke–Loewenstein tumour) and verrucous squamous carcinoma of the perianal and anorectal region: a continuous precancerous spectrum? Histopathology 1985;9:155–169.
Trombetta LJ, Place RJ . Giant condyloma acuminatum of the anorectum: trends in epidemiology and management: report of a case and review of the literature. Dis Colon Rectum 2001;44:1878–1886.
Chiesa-Vottero A, Dvoretsky PM, Hart WR . Histopathologic study of thin vulvar squamous cell carcinomas and associated cutaneous lesions: a correlative study of 48 tumors in 44 patients with analysis of adjacent vulvar intraepithelial neoplasia types and lichen sclerosus. Am J Surg Pathol 2006;30:310–318.
Iaconis L, Hyjek E, Ellenson LH, et al. p16 and Ki-67 immunostaining in atypical immature squamous metaplasia of the uterine cervix: correlation with human papillomavirus detection. Arch Pathol Lab Med 2007;131:1343–1349.
Liegl B, Regauer S . P53 immunostaining in lichen sclerosus is related to ischaemic stress and is not a marker of differentiated vulvar intraepithelial neoplasia (d-VIN). Histopathology 2006;48:268–274.
Chulvis do Val IC, Almeida Filho GL, Valiante PM, et al. Vulvar intraepithelial neoplasia p53 expression, p53 gene mutation and HPV in recurrent/progressive cases. J Reprod Med 2004;49:868–874.
Rolfe KJ, MacLean AB, Crow JC, et al. TP53 mutations in vulval lichen sclerosus adjacent to squamous cell carcinoma of the vulva. Br J Cancer 2003;89:2249–2253.
Acknowledgements
We thank J Lindeman and Labo Bio-Medical Products BV (Rijswijk, The Netherlands) for providing the RHA strips.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
de Koning, M., Quint, W. & Pirog, E. Prevalence of mucosal and cutaneous human papillomaviruses in different histologic subtypes of vulvar carcinoma. Mod Pathol 21, 334–344 (2008). https://doi.org/10.1038/modpathol.3801009
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3801009
Keywords
This article is cited by
-
MTA1 as negative prognostic marker in vulvar carcinoma
Journal of Cancer Research and Clinical Oncology (2023)
-
Evidence of the causal role of human papillomavirus type 58 in an oropharyngeal carcinoma
Virology Journal (2013)
-
Comprehensive analysis of human papillomavirus prevalence and the potential role of low-risk types in verrucous carcinoma
Modern Pathology (2012)
-
HPV-assoziierte Veränderungen an Vulva und Vagina
Der Pathologe (2011)
