
Abstract
Renal cell carcinomas in young patients constitute a morphologically and genetically heterogeneous group. Twenty percent belong to the newly recognized Xp11.2 translocation-associated family and rare tumors arise from nephroblastoma. Aberrant Wnt signaling through β-catenin mutation has been implicated in nephroblastoma pathogenesis and has been found to synergize with WT1 mutations. To characterize Wnt signaling activity in renal cell carcinomas in young patients, we gathered 34 tumors (three clear cell, ten Xp11.2 translocation associated, five papillary, two chromophobe, two collecting duct, one neuroblastoma associated, eight unclassified renal cell carcinomas, and three carcinomas combined with nephroblastoma) from patients less than 22 years. Expression of β-catenin, its homologue γ-catenin, and of WT1 was assessed by immunohistochemistry in 30 tumors, and sequence analysis of CTNNB1, CTNNG1, and WT1 genes was performed in 25 tumors. Cytoplasmic β-catenin accumulation was demonstrated in two papillary carcinomas, one neuroblastoma-associated carcinoma, and two carcinomas arising from nephroblastoma. The pattern of γ-catenin expression paralleled that of β-catenin but its signal intensity was lower in 22, equal in 7, and stronger only in 1 tumor, respectively. Four tumors showed nuclear WT1 expression. One Xp11.2 translocation-associated carcinoma presented a rare intronic CTNNB1 single nucleotide polymorphism and cytoplasmic β-catenin accumulation. There were no further CTNNB1 or CTNNG1 sequence alterations. A WT1 mutation was found in the nephroblastoma component of a carcinoma arising from nephroblastoma. These findings suggest Wnt signaling pathway activation only in a minority of renal cell carcinomas in young patients. CTNNB1 mutations are rare events. Cytoplasmic β-catenin accumulation in an Xp11.2-associated carcinoma suggests potential interaction of Wnt signaling components with microphthalmia transcription factor family also in Xp11.2 translocation carcinomas. WT1 mutation in the nephroblastoma component of a mixed-type renal cell carcinoma provides direct evidence for clonal independence of nephroblastoma and carcinoma components in this exceptional tumor.
Similar content being viewed by others

Main
Proliferation-associated signaling pathways are temporarily active during embryogenesis and direct rapid growth of developing organs. As soon as organ development is completed, these pathways are usually shut down. However, in some cells like adult stem cells, the same pathways may remain active for adult tissue maintenance. Unrestricted activation of such signaling pathways has been implicated in human cancer pathogenesis, both in children and in adults. Wnt signaling is of central importance for the development of many organs and has been implicated in tumor pathogenesis at diverse sites such as skin,1 brain,2 liver,3 and prostate.4 Wnt signaling is initiated by binding of the secreted Wnt proteins to their cognate receptors. β-Catenin is one of the critical Wnt receptor targets.5 β-Catenin phosphorylation by glycogen synthase kinase 3β is catalyzed by simultaneous binding of the APC protein and normally targets β-catenin to rapid ubiquitin-mediated degradation.5 Activation of the Wnt signaling pathway, however, abolishes β-catenin phosphorylation, leads to β-catenin stabilization and subsequent nuclear translocation. Within the nucleus, β-catenin interacts with members of the lymphoid-enhancing factor 1 (LEF-1)/TCF family and generates a functional transcription factor complex that causes transcriptional activation of downstream target genes including CMYC and CCND1.5 Similarly, TFE3/microphthalmia-associated transcription factor interacts with LEF-1 as mediator of the Wnt signaling pathway.6
Renal cell carcinoma in young patients is rare and differs substantially from its adult counterpart in morphology, genetic background, and outcome.7, 8, 9 In children and adolescents, tumors associated with the Xp11.2 translocation and TFE3 overexpression constitute a major proportion of renal cell carcinomas.7, 8, 10 These Xp11.2 translocation-associated carcinomas as well as carcinomas associated with neuroblastoma form new entities now included in the WHO renal cell carcinoma classification.11 Exceptionally, renal cell carcinomas in young patients may arise from metanephric adenofibroma or nephroblastoma.8, 12 Nephroblastoma, the most common malignant renal tumor in childhood, is thought to arise from pluripotent precursor cells within metanephric blastema. A significant subset of nephroblastomas shows constitutive activation of Wnt signaling caused by β-catenin gene (CTNNB1) mutations.13 Of note, CTNNB1 mutations almost invariably occur in nephroblastomas harboring concomitant WT1 mutations.14
As independent evidence from transgenic mouse models recently showed that mice lacking the Apc gene specifically in the kidney are prone to the development of renal cell carcinomas,15 we chose to investigate the functional role of Wnt signaling in renal cell carcinomas of young patients. We studied immunohistochemical expression of β-catenin, γ-catenin, and WT1 in 30 renal cell carcinomas, combined with sequencing analysis for activating mutations of exons 3, 7, and 8 of CTNNB1, exon 2 of its homologous γ-catenin gene CTNNG1, and for WT1 gene mutations of exons 7, 8, 9, and 10, encoding the zinc-finger region of WT1, by genomic amplification and direct sequencing in 25 renal cell carcinomas, including three carcinomas arising in the context of nephroblastoma.
Materials and methods
Renal Cell Carcinomas
Thirty-four renal cell carcinomas of 34 patients were compiled from the archives of the Pediatric Tumor Registry, University Hospital Schleswig-Holstein Campus Kiel, Germany, and the Institute of Pathology, University Hospital Basel, Switzerland. Only tumors with available paraffin blocks were included. The renal cell carcinomas were classified according to the 2004 WHO classification as described previously (Table 1).8, 11 There were three clear cell, ten Xp11.2 translocation-associated, five papillary, two chromophobe, two collecting duct, and eight unclassified renal cell carcinomas. The 10 Xp11.2 translocation-associated carcinomas were classified based on immunohistochemical nuclear TFE3 overexpression in all 10 tumors. Immunohistochemical TFE3 overexpression has previously been shown to constitute a highly sensitive and specific assay for neoplasms bearing a TFE3 gene fusion.10, 16 One carcinoma occurred after neuroblastoma and three carcinomas arose in combination with nephroblastoma. Twenty-nine tumors were part of our previous study on morphologic and molecular analysis and were reviewed by JE, IL, DH, HM, PA, ML, and EB,8 five tumors were added to the study set and reviewed by three authors (IL, PA, and EB).
Immunohistochemistry
Immunohistochemistry for β-catenin, γ-catenin, and WT1 was performed on sections of a pediatric renal cell carcinoma paraffin tumor tissue array. The paraffin tumor array was constructed on a recipient paraffin block with punches of 0.6 mm diameter from donor paraffin blocks as described previously.17 Two punch cylinders per tumor were sampled. Of the three renal cell carcinomas arising from Wilms tumor, both renal cell carcinoma and Wilms tumor components were included. A total of 30 tumors were represented on the tumor array. Normal renal parenchyma was present and included in 20 patients. Immunohistochemical reactions including antigen retrieval were performed on a Vision Biosystems Bond X Immunostainer according to the manufacturer's protocol. Primary antibodies were as follows: β-catenin 1:200 (Transduction, Becton Dickinson Biosciences, San Jose, CA, USA); γ-catenin 1:50 (Novocastra, Newcastle upon Tyne, UK); and WT1 1:50 (Novocastra, Newcastle upon Tyne, UK). The ABC chromogen detection method was applied. For each immunohistochemical reaction, a negative control without primary antibody was included. A nephroblastoma served as positive control for WT1 immunohistochemistry. For β-catenin and γ-catenin, normal renal tissue was used as internal positive control. Evaluation of immunohistochemical reactivity was performed in a similar way as proposed by Kim et al18 and as detailed in the Results section below.
DNA Preparation, PCR Amplification, Sequencing
Sequencing of CTNNB1, CTNNG1, and WT1 genes was performed on 25 tumors (Table 1). For preparation of sample DNA, five 0.6 mm paraffin tissue punch cylinders were removed from identical matched tumor regions from the original paraffin tissue blocks of each of the 25 tumors. In two renal cell carcinomas arising from Wilms tumor, renal cell carcinoma and Wilms tumor components were sampled separately for sequencing analysis. DNA extraction from formalin-fixed, paraffin-embedded tissue was performed with a QIAamp® DNA Mini kit from Qiagen following the manufacturer's protocol. Exons 3, 7, and 8 of CTNNB1, exon 2 of CTNNG1, and exons 7, 8, 9, and 10 of WT1 were amplified by PCR (primer sequences in Table 2). PCR cycling conditions were: 2 mM MgCl2 and 1 U Platinum-Taq polymerase (Invitrogen), initial denaturation 5 min 94°C, 40 denaturation cycles 30 s 94°C, 30 s annealing 52–60°C, 30 s elongation 72°C, and final elongation 7 min 72°C. PCR fragments were purified using High Pure PCR Purification kit (Roche Diagnostics) essentially as recommended by the manufacturer. Sequencing reactions were set up with 30 ng purified PCR fragment template and 10 pmol sequencing primer in 10 μl total reaction volume following a dye terminator protocol (Big Dye, Applied Biosystems) on an ABI Prism 310 DNA Sequencer (Applied Biosystems). Sequencing primers were identical to amplification primers. Sequence alterations were verified by sequencing both DNA strands and by analyzing an independently generated PCR amplicon.
Results
Patients and Tumors
A total of 34 renal cell carcinomas were investigated. Patient age ranged between 4 and 22 years, median age 12 years. Gender ratio was equal with 17 male to 17 female patients. Tumor classification according to the 2004 WHO classification is shown in Table 1. There were three clear cell renal cell carcinomas, ten Xp11.2 translocation-associated renal cell carcinomas, five papillary renal cell carcinomas, two chromophobe carcinomas, two collecting duct carcinomas, one neuroblastoma-associated renal cell carcinoma, three renal cell carcinomas arising from Wilms tumors, and eight unclassified renal cell carcinomas.
β-Catenin, γ-Catenin, and WT1 Immunohistochemistry
Immunohistochemical results are summarized in Table 1. To detect alterations in β- and γ-catenin expression in renal cell carcinomas, immunohistochemical reactivity was studied and graded according to signal location and intensity in comparison to epithelia of normal renal tubules in a similar way as proposed by Kim et al18 Normal renal tissue was present in 20 tumors. In normal renal tissue, β-catenin expression was strong and membranous in epithelia of collecting ducts and Henle loops, whereas proximal and distal tubules showed predominantly cytoplasmic expression of moderate intensity. There was no nuclear β-catenin expression in any of the normal renal compartments. γ-Catenin expression in normal renal tissue was similar to β-catenin, but slightly weaker. Membranous, cytoplasmic, and nuclear expression was distinguished in β-catenin and γ-catenin reactions. Nuclear WT1 expression was separated from cytoplasmic WT1 expression. Signal intensity was graded from + to +++ in a semiquantitative manner (see Table 1). Membranous β-catenin expression was regarded as strong/+++ if similar to the strongest signal in normal kidney in collecting duct epithelia, moderate/++ or weak/+ if there was reduced signal intensity. Cytoplasmic β-catenin expression was regarded as moderate/++ if similar to proximal renal tubules, strong/+++ if signal intensity was stronger, and weak/+ if signal intensity was reduced, respectively. Grading of γ-catenin expression was performed in a similar manner. Immunohistochemical staining was homogeneous in all tumor cells in the majority of tumors, with minor foci of less than 10% of tumor cells with divergent expression, termed ‘focal’ expression in Table 1. Immunohistochemical data were analyzable in 30 tumors represented on the paraffin tissue tumor array. Strong/+++ membranous β-catenin expression was present in the majority of tumors (19 out of 30). Interestingly, in 2 of 4 papillary type II renal cell carcinomas, membranous β-catenin expression was located to the basolateral cell membrane. Strong/+++ cytoplasmic β-catenin expression, exceeding expression in epithelia of normal renal tubules, was found in 6 of 30 tumors: one translocation-associated renal cell carcinoma (Figure 1), two papillary type II renal cell carcinomas (Figure 2), one neuroblastoma-associated renal cell carcinoma, and two renal cell carcinomas arising from Wilms tumors (Figures 3 and 4). Strong nuclear β-catenin expression was not found in any tumor. Five tumors showed focal or diffuse weak/+ nuclear expression, among these the nephroblastoma component of a renal cell carcinoma arising from nephroblastoma (tumor 26) and the neuroblastoma-associated renal cell carcinoma (tumor 23).

Xp11.2-translocation-associated carcinoma (tumors 4 and 5). (a, b) Xp11.2 translocation-associated renal cell carcinoma with ample ‘voluminous’ cytoplasm and solid growth pattern (H&E × 400; tumor 4). Strong membranous, weak cytoplasmic, no nuclear β-catenin expression (immunohistochemistry for β-catenin × 400). (c, d) Xp11.2 translocation-associated renal cell carcinoma with characteristic psammoma bodies and tubular growth pattern; tumor with CTNNB1 single nucleotide polymorphism (H&E × 400; tumor 5). Strong membranous, and cytoplasmic, no nuclear β-catenin expression (immunohistochemistry for β-catenin × 400).

Chromophobe carcinoma, clear cell carcinoma, papillary carcinoma. (a, b) Chromophobe carcinoma. H&E × 400. Strong membranous, weak cytoplasmic β-catenin expression (immunohistochemistry for β-catenin × 400; tumor 20). (c, d) Clear cell carcinoma. H&E × 400. Moderate membranous β-catenin expression (immunohistochemistry for β-catenin × 400; tumor 2). (e, f) Papillary renal cell carcinoma type II. Strong membranous basolateral β-catenin expression and moderate cytoplasmic accumulation (immunohistochemistry for β-catenin × 400; tumor 18).

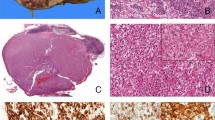
Renal cell carcinoma arising from nephroblastoma (tumor 25). (a, b) Metanephric adenoma component composed of small tumor cells with little cytoplasm forming sheets, nests, and tubular aggregates. Focal distinct membranous, moderate cytoplasmic, and weak nuclear β-catenin expression (immunohistochemistry for β-catenin × 400). (c, d) Solid tumor component with larger nuclei and broader cytoplasmic rim consistent with renal cell carcinoma component. Strong membranous β-catenin expression (immunohistochemistry for β-catenin × 400). (e, f) Papillary tumor component metastatic to a regional lymph node, consistent with papillary renal cell carcinoma. Strong membranous, cytoplasmic, and weak nuclear β-catenin expression (immunohistochemistry for β-catenin × 400).

Renal cell carcinoma arising from nephroblastoma (tumor 26). (a) Solid sheets of small tumor cells with narrow cytoplasmic rim consistent with nephroblastoma component. H&E × 400. (b) Solid and tubular growth pattern of larger tumor cells with ample eosinophilic cytoplasm and large nuclei with prominent nucleoli, consistent with renal cell carcinoma. H&E × 400. (c, d) Focal membranous β-catenin expression in the nephroblastoma component, strong membranous, cytoplasmic, and weak nuclear expression in the component consistent with renal cell carcinoma (immunohistochemistry for β-catenin × 400). (e, f) Focal cytoplasmic WT1 expression in the nephroblastoma component, nuclear expression in the renal cell carcinoma component (immunohistochemistry for WT1 × 400).
In 5 of 8 Xp11 translocation-associated and 2 of 3 clear cell renal cell carcinomas, β-catenin expression was membranous and strong (Figure 1), whereas 1 of 2 collecting duct and unclassified chromophobe-like carcinomas, respectively, was weakly staining or negative. The unclassified vacuolar lipoid renal cell carcinoma was negative for β-catenin.
γ-Catenin immunohistochemistry paralleled that of β-catenin with slightly weaker intensity (Table 1). Exceptions were one Xp11.2 translocation-associated renal cell carcinoma with strong/+++ membranous γ-catenin expression in contrast to weak/+ membranous β-catenin expression, and two papillary renal cell carcinomas with equally strong β- and γ-catenin expression. Similar to β-catenin, γ-catenin showed a basolateral expression pattern in papillary renal cell carcinoma type II. Strong/+++ membranous γ-catenin expression was also observed in the following: one post-neuroblastoma renal cell carcinoma, the renal cell carcinoma components of one renal cell carcinoma, the nephroblastoma component of two renal cell carcinomas arising from nephroblastoma, respectively, and in two unclassified renal cell carcinomas.
Nuclear WT1 expression was observed in the nephroblastoma components of two renal cell carcinomas arising from nephroblastoma (Figure 4), in one renal cell carcinoma component arising from nephroblastoma, and in one unclassified renal cell carcinoma with prominent cytoplasmic vacuolization. Furthermore, predominantly weak-to-moderate cytoplasmic positivity for WT1 was observed in eight tumors. All other carcinomas were WT1 negative.
CTNNB1, CTNNG1, and WT1 Sequencing
Mutational activation of CTNNB1 might be involved in the development of childhood renal cell carcinomas. Earlier studies have identified exon 3 encoding the consensus GSK-3β phosphorylation sites of β-catenin as the major CTNNB1 mutational target region in colon,5 skin,2 and prostate4 cancer, and also in a variety of childhood tumors, including hepato-, nephro-, medullo-, and pancreatoblastomas (reviewed in Koesters and von Knebel Doeberitz19). More recently, evidence has been provided that mutations of exon 7 and 8 are functionally equal to exon 3 mutations in at least a subset of Wilms tumors.20 We therefore amplified CTNNB1 exons 3, 7, and 8 and adjacent intron regions. PCR products were submitted to direct sequencing. Our sequence analysis yielded a rare CTNNB1 intron 7 single nucleotide polymorphism21 in immediate vicinity of the splice acceptor site in a renal cell carcinoma of TFE3 translocation-associated subtype (tumor 5; Figure 5). The other 24 renal cell carcinomas including those arising from nephroblastoma showed wild-type CTNNB1 sequences.

(a) CTNNB1 and WT1 sequencing results. 5ACTNNB1 intron 7 and WT1 exon 7 sequences: electropherograms of CTNNB1 intron 7 before the splice acceptor site (left panel), wild-type control sample (top), direct sequencing of PCR fragments derived from tumor 5 (bottom). A rare single nucleotide polymorphism (C/G) was present in this tumor. Electropherograms of WT1 codon 346 (right panel), wild-type control sample (top), and direct sequencing of PCR fragments derived from the nephroblastoma component of combination tumor 26. The heterozygous mutation AAG346AGG results in replacement of lysine 346 by arginine in the first zinc-finger of WT1. (b) CTNNB1 single nucleotide polymorphism and WT1 mutation: the CTNNB1 intron 7 single nucleotide polymorphism in a Xp11.2 translocation-associated carcinoma (tumor 5) is located 17 nucleotides before the splice acceptor site (top panel). The WT1 codon 346 mutation of the nephroblastoma component of the combination tumor (tumor 26) results in replacement of lysine by arginine (red) at the base of the first zinc-finger (bottom panel). Asterisks indicate mutations previously found in Denys–Drash patients.35
Similar to β-catenin, γ-catenin has important functions in cell adhesion and in Wnt signal transduction.22, 23 γ-Catenin is regulated by APC,24 and oncogenic CTNNG1 mutations have been identified in vitro24 as well as in cancers of the digestive tract.25 We therefore amplified the corresponding region within exon 2 of CTNNG1, which harbors the putative GSK3β phosphorylation sites of γ-catenin. In this case, we did not find a CTNNG1 mutation in any of the 25 tumors with available genomic DNA.
Renal cell carcinomas in children may exceptionally arise in combination with nephroblastoma, the most common malignant renal neoplasm in childhood. Three renal cell carcinomas in this series were judged to arise from nephroblastoma. As WT1 mutations frequently occur in nephroblastomas26, 27 and are highly associated with CTNNB1 mutations in those cancers,13, 14 we also investigated whether WT1 mutations might contribute to the development of renal cell carcinoma in young patients. We amplified exons 7, 8, 9, and 10 encoding the four zinc-fingers of WT1 in which the majority of WT1 mutations have been found.27 In two of the three combined tumors, we investigated renal cell carcinoma and nephroblastoma components separately. In 1 of the 25 tumors analyzed, we detected a single nucleotide exchange (A → G) at codon 346, resulting in substitution of a lysine residue by arginine in the first zinc-finger of WT1 (Figure 5). This mutation was found in a heterozygous state only in the nephroblastoma component, but not the renal cell carcinoma component of a renal cell carcinoma arising from nephroblastoma. None of the other 24 tumors showed any WT1 sequence alteration.
Discussion
To characterize Wnt pathway activity in renal cell carcinomas in young patients, we have analyzed immunohistochemical β-catenin, γ-catenin, and WT1 expression in 30 and CTNNB1, CTNNG1, and WT1 gene sequences in 25 pediatric and adolescent renal cell carcinomas. Cytoplasmic accumulation of β-catenin was observed in 6 out of 30 tumors in our current study: 1 of 8 Xp11.2 translocation-associated carcinoma, 2 of 4 papillary carcinomas, 1 post-neuroblastoma carcinoma, and 2 of 3 renal cell carcinomas associated with nephroblastoma. These findings are in contrast to previous investigations in renal carcinomas in adult patients by Kim et al,18 who reported cytoplasmic β-catenin accumulation in 5 of 22 clear cell carcinomas but none of 10 papillary carcinomas.
The Xp11.2 translocation-associated carcinoma revealed a rare CTNNB1 single nucleotide polymorphism located in close proximity to the splice acceptor site of intron 7. Conceivably, this sequence variant may lead to elevated β-catenin protein levels by either increasing the efficiency of splicing or by causing alternative splicing with the result of a stabilized β-catenin protein. In support of this hypothesis, we found strong cytoplasmic β-catenin expression in this tumor. In addition, weak nuclear translocation of β-catenin was present in 3 of the 6 renal cell carcinomas with cytoplasmic accumulation. These findings suggest a potential contribution of Wnt pathway signaling activation in pediatric renal cell carcinoma pathogenesis.
γ-Catenin expression paralleled that of β-catenin, but was usually weaker in most tumors. Elevated levels of γ-catenin expression were also found in some tumors demonstrating only low levels of β-catenin, suggesting independent regulation of both catenin proteins.
Of note, in the recently described Xp11.2 translocation-associated renal cell carcinomas, TFE3 protein is overexpressed as a result of TFE3 fusion.8, 10 TFE3 is a member of the microphthalmia family of transcription factors. This transcription factor family is comprised of MITF, TFE3, TFEB, and TFEC, and all family members share a highly homologous basic helix–loop–helix leucine zipper (bHLHzip) DNA binding and dimerization domain.28 These proteins homo- or heterodimerize in all combinations and bind identical DNA elements, suggesting that they may activate common downstream targets.29 Elegant knockout studies have demonstrated functional redundancy of MITF and TFE3 in modulating murine osteoclast development.30 In a cotransfection experiment, it was clearly shown that in the context of translocation-associated renal cell carcinoma, MITF can functionally replace translocated TFE3 in promoting oncogenic growth.31 MITF is a Wnt signaling downstream target, and in mammalian cells, Wnt3a was also shown to induce MITF expression.32 Of interest, interactions between the MITF family and the Wnt signaling pathway are increasingly being recognized for some tumors. In particular, in human melanoma, β-catenin is deregulated via aberrant nuclear accumulation and is a potent growth mediator for melanoma cells dependent on its downstream target MITF, so that β-catenin regulation of MITF expression represents a pathway with significant influence on neoplastic growth.33 In addition, interaction of the TFE3/microphthalmia transcription factor family has been implied with LEF-1 as mediator of the Wnt pathway in a cell culture approach.6 Simultaneous CTNNB1 gene sequence variant, cytoplasmic accumulation of β-catenin, and overexpression of TFE3 in one Xp11.2 translocation-associated renal cell carcinoma in our series may support a potential interaction of the Wnt pathway with the TFE3/microphthalmia transcription factor family in the pathogenesis of a subset of renal cell carcinomas in young patients and may thus suggest a complementary pathway for oncogenic TFE3 upregulation in these tumors. However, as this has been observed in only one tumor in our study, this pathway is probably of minor importance in renal carcinogenesis for young patients.
The von Hippel-Lindau (VHL) tumor suppressor gene product has been implied in suppression of oncogenic β-catenin signaling in renal carcinoma cells in adults.34 However, in contrast to adult sporadic clear cell renal cell carcinoma with a high percentage of VHL gene mutations and loss of heterozygosity of the VHL gene region, in renal cell carcinoma in young patients, VHL gene mutations are absent.8 In this regard, the lack of β-catenin accumulation especially in the clear cell renal cell carcinomas in our current study is in accordance with our previous demonstration of the absence of VHL mutations and rarity of VHL alterations in renal cell carcinomas in young patients.
Interestingly, the WT1 mutation found in tumor 26 was present only in the Wilms tumor component, but was absent in the renal cell carcinoma component. The importance of this finding is two-fold. First, it provides direct evidence that this mutation has been acquired somatically as a tumor-associated event, and that it is not a rare polymorphism. Second, because the WT1 mutation serves as a powerful clonal marker, it proves the independent origin of both tumors. In particular, in this specific tumor, it can be excluded that the renal cell carcinoma has arisen from the Wilms tumor simply by tumor progression. It is open to further investigations to examine if this is true for all these rare mixed-type renal cell cancers. Although the WT1 mutation was found only in a heterozygous state in the tumor, we cannot exclude that the second WT1 allele has also been inactivated outside the zinc-finger region, at a position not covered by our analysis. Furthermore, it is known in patients with Denys–Drash syndrome that some WT1 mutations may even exert dominant negative functions over the wild-type allele.35 Hence, the present mutation (lysine 346 arginine) found at the base of the first zinc-finger of WT1 is likely to be of functional importance.
In summary, activation of the Wnt signaling pathway may occur in a subset of renal cell carcinomas in young patients, but CTNNB1 mutations are rare events. Cytoplasmic β-catenin accumulation in the presence of TFE3 overexpression may suggest potential interaction of the Wnt signaling pathway with members of the microphthalmia family of transcription factors also in Xp11 translocation-associated renal cell carcinoma, but this pathway is probably of minor importance in renal carcinogenesis for young patients.
References
Robbins PF, El-Gamil M, Li YF, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med 1996;183:1185–1192.
Zurawel RH, Chiappa SA, Allen C, et al. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res 1998;58:896–899.
de La Coste A, Romagnolo B, Billuart P, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA 1998;95:8847–8851.
Voeller HJ, Truica CI, Gelmann EP . Beta-catenin mutations in human prostate cancer. Cancer Res 1998;58:2520–2523.
Clevers H . Wnt/β-catenin signaling in development and disease. Cell 2006;127:469–480.
Yasumoto K, Takeda K, Saito H, et al. Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J 2002;21:2703–2714.
Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor previously included among renal cell carcinomas of children and adolescents. Am J Pathol 2001;159:179–192.
Bruder E, Passera O, Harms D, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol 2004;28:1117–1132.
Selle B, Furtwaengler R, Graf N, et al. Population-based study of renal cell carcinoma in children in Germany, 1980–2005: more frequently localized tumors and underlying disorders compared with adult counterparts. Cancer 2006;107:2906–2914.
Argani P, Lal P, Hutchinson B, et al. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol 2003;27:750–761.
Eble JN, Sauter G, Epstein JI, et al (eds). Tumours of the Kidney. In: WHO Classification of Tumours: Tumours of the Urinary System and Male Genital Organs. IARC Press: Lyon, France, 2004, pp 12–43.
Arroyo MR, Green DM, Perlman EJ, et al. The spectrum of metanephric adenofibroma and related lesions: clinicopathologic study of 25 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 2001;25:433–444.
Koesters R, Ridder R, Kopp-Schneider A, et al. Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res 1999;59:3880–3882.
Maiti S, Alam R, Amos CI, et al. Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 2000;60:6288–6292.
Sansom OJ, Griffiths DF, Reed KR, et al. Apc deficiency predisposes to renal carcinoma in the mouse. Oncogene 2005;24:8205–8210.
Argani P, Olgac S, Tickoo S, et al. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol 2007;31:1149–1160.
Kononen J, Bubendorf L, Kallioniemi A, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998;4:844–847.
Kim Y-S, Kang YK, Kim JB, et al. Catenin expression and mutational analysis in renal cell carcinomas. Pathol Internat 2000;50:725–730.
Koesters R, von Knebel Doeberitz M . The Wnt signaling pathway in solid childhood tumors. Cancer Lett 2003;198:123–138.
Li CM, Kim CE, Margolin AA, et al. CTNNB1 mutations and overexpression of Wnt/beta-catenin target genes in WT1-mutant Wilms’ tumors. Am J Pathol 2004;165:1943–1953.
National Center for Biotechnology Information (NCBI). Single Nucleotide Polymorphism. rs2293302. Build 13. Revision May 2006 (online). Available at: http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2293302;Accessed 13 June 2007.
Cadigan KM, Nusse R . Wnt signaling: a common theme in animal development. Genes Dev 1997;11:3286–3305.
Barth AI, Näthke IS, Nelson WJ . Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol 1997;9:683–690.
Kolligs FT, Kolligs B, Hajra KM, et al. Gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev 2000;14:1319–1331.
Caca K, Kolligs FT, Ji X, et al. Beta- and gamma-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell Growth Differ 1999;10:369–376.
Koesters R, Adams V, Hassam S . Mutational analysis of the tumor suppressor gene WT1: detection of a novel homozygous point mutation in sporadic unilateral Wilms tumor. Int J Oncol 1995;7:1103–1107.
Varanasi R, Bardeesy N, Ghahremani M, et al. Fine structure analysis of the WT1 gene in sporadic Wilms tumors. Proc Natl Acad Sci USA 1994;91:3554–3558.
Steingrimsson E, Copeland NG, Jenkins NA . Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 2004;38:365–411.
Hemesath TJ, Steingrimsson E, McGill G, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev 1994;8:2770–2780.
Steingrimsson E, Tessarollo L, Pathak B, et al. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc Natl Acad Sci USA 2002;99:4477–4482.
Davis IJ, Kim JJ, Ozsolak F, et al. Oncogenic MITF dysregulation in clear cell sarcoma: defining the MiT family of human cancers. Cancer Cell 2006;9:473–484.
Takeda K, Yasumoto K, Takada R, et al. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J Biol Chem 2000;275:14013–14016.
Widlund HR, Horstmann MA, Price ER, et al. Catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J Cell Biol 2002;158:1079–1087.
Peruzzi B, Gagani A, Bottaro DP . The von Hippel-Lindau tumor suppressor gene product represses oncogenic β-catenin signaling in renal carcinoma cells. Proc Natl Acad Sci 2006;103:14531–14536.
Coppes MJ, Campbell CE, Williams BR . The role of WT1 in Wilms tumorigenesis. FASEB 1993;7:886–895.
Acknowledgements
We are deeply grateful to John Eble for his major contribution to consensus classification of these renal cell carcinomas and review of the manuscript. We cordially thank Marc Ladanyi for collaborative interpretation of the CTNNB1 sequencing results and review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
This study was supported by the Basel Cancer League. The authors have no conflict of interest to disclose.
Rights and permissions
About this article
Cite this article
Bruder, E., Moch, H., Ehrlich, D. et al. Wnt signaling pathway analysis in renal cell carcinoma in young patients. Mod Pathol 20, 1217–1229 (2007). https://doi.org/10.1038/modpathol.3800957
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800957
