
Abstract
The diagnosis of viral myocarditis remains difficult and generally depends on clinical and histologic criteria. Viral cultures and serology are often unrewarding with low yields. The purpose of this study was to analyze the usefulness of PCR in the rapid diagnosis of myocarditis in children. PCR was used to analyze 120 myocardial tissue samples from 60 cases of sudden infant death syndrome (SIDS) and 56 myocardial tissue samples from 36 cases with well-known causes of sudden death (11 children younger than 1 year and 25 children 1–10 years old). The myocardial tissue samples were evaluated for the presence of enteroviruses and parvovirus B19 using PCR primers designed to consensus and unique sequences of these viral genomes. Enteroviruses could be detected in 14 cases of SIDS, whereas the detection of enteroviral nucleic acid within the control group was negative. Seven cases with myocardial infection caused by parvovirus B19 were found in the SIDS group. The detection of parvoviruses in the control group of the 11 children younger than 1 year was negative, whereas 3 positive cases of parvoviruses could be detected in the control group of children from 1 to 10 years old. In the myocardial sample of one SIDS case, both enteroviruses and parvovirus B19 could be detected. Our results emphasize the importance of modern molecular biologic methods in cases of sudden infant death even when conventional histologic examination revealed no serious findings in heart muscle tissue.
Similar content being viewed by others

Introduction
Infants dying suddenly and unexpectedly in whom the autopsy fails to establish the cause of death have been the subject of a number of studies attempting to discover an infectious agent that may explain their deaths. Rambaud et al (1992) identified signs of myocarditis using conventional histologic methods in 24 (16.8%) of 143 infants who died suddenly and unexpectedly. In a recent series of pediatric myocarditis cases, enterovirus, cytomegalovirus, and adenovirus together accounted for 63% of total cases for whom an etiologic agent could be established by genome amplification. These studies, together with epidemiologic aspects, point to an inflammatory process in cases of sudden infant death syndrome (SIDS) and suggest that new molecular-pathologic methods could lead to an higher incidence of myocarditis. Recent studies also indicate that PCR rapidly detects the presence of viral nucleic acid in a variety of infected tissues and body fluids (Bendig et al, 2001; Lau, 1994; Lozinski et al, 1994; Murry et al, 2001)
Enteroviruses are the most common agents of myocarditis. Viral proteins are synthesized as a polyprotein, subsequently cleaved to produce the four capsid proteins VP 1 through VP 4. RT-PCR as well as in situ hybridization and serologic studies revealed an association between enterovirus infection, especially the cardiotropic coxsackieviruses group B, and myocarditis (Jin et al, 1990). The family Parvoviridae is a group of DNA viruses that are known to infect both animals and humans. Parvovirus B19 is the only known human pathogen in this family of viruses (Cherry, 1999). The infection is relatively common in humans, and nearly 50% of the population have detectable antibodies by the age of 15 years, increasing to 90% in the elderly (Cohen and Buckley, 1988). Human parvoviral infection usually causes asymptomatic infections or erythema infectiosum (“fifth disease“) accompanied by a characteristic facial rash.
To investigate whether viruses known to cause myocarditis are associated with the subset of infants dying suddenly and unexpectedly, we performed PCR assays for the presence of genomic sequences from enteroviruses and parvovirus B19 in paraffin-embedded heart tissues.
Results
The results of the presence of enteroviruses and parvovirus B19 are listed in Table 1. We investigated two different heart regions from each individual (septum interventriculare cranial and left ventricle posterior wall cranial). Enteroviruses could be detected in 14 of 60 cases of suspected SIDS, whereas 8 of 60 cases were positive for parvovirus B19. The detection of enteroviral acid and parvovirus B19 of the control group (children younger than 1 year) was negative, whereas the parvovirus B19 genome could be detected in three cases of the comparison group from older children (1–10 years old). In the myocardial sample of Case 8, both enteroviruses and parvovirus B19 were found. All positive samples for enteroviruses and parvovirus B19 were amplified in duplicate, starting from the first extraction step. Therefore false-positive results can be excluded.
In all SIDS cases (except Case 8), the myocardial samples revealed no signs of myocarditis according to the Dallas criteria using conventional histologic stainings. The PCR-based detection of enteroviruses correlated in seven cases (Cases 2–6, 8, and 19) with the detection of enteroviral capsid protein VP1 (data not shown). In addition, we performed enteroviruses and parvovirus B19 (RT)-PCR of spleen and liver tissue concurrently with that of myocardial samples, and the spleen and the liver were negative in the face of a positive cardiac (RT-)PCR.
Enterovirus and parvovirus B19 (RT-)PCR results are shown in Figures 1 and 2. All enteroviral and parvovirus B19-positive PCR products were analyzed by direct sequencing. Sequence comparison was performed by a BLAST search of GenBank databases. The PCR primers for enteroviruses and parvovirus B19 was designed to consensus and unique sequences of these viral genomes. Therefore the sequencing results showed homology to the comparison sequence of enteroviruses and parvovirus B19.

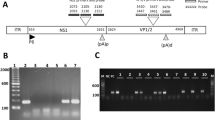
Enterovirus diagnostic (110-bp fragment; Accession No. AF177911). Amplification of the products of RT-PCR. The products were subjected to electrophoresis on an 8% polyacrylamide gel, and DNA fragments were detected by silver staining. Lanes: 1 and 6, 50-bp ladder; 2, negative control; 3, positive control; 4, extraction control; 5, Case 8.

Parvovirus B19 diagnostic (120-bp fragment; Accession No. AB030694). PCR products after nested PCR were subjected to electrophoresis on an 8% polyacrylamide gel, and DNA fragments were detected by silver staining. Lanes: 1 and 6, 50-bp ladder; 2, negative control; 3, positive control; 4, extraction control; 5, Case 7.
Discussion
There are no data concerning the distribution of myocarditis in adults during the year. Grady and Costanzo-Nordin (1989) emphasize that the distribution of myocarditis may vary with age, sex, and season of the year.
Recent studies concerning SIDS cases as well as older children detected different viruses in cases of myocarditis using molecular methods to search for the genome sequences of enteroviruses (Bendig et al, 2001; Lau, 1994), adenovirus (Lozinski et al, 1994), parvovirus B19 (Murry et al, 2001), Epstein-Barr virus, and human herpes viruses in clinical or autopsy samples (Towbin et al, 1993). Some authors speculate that there are cases of myocarditis misdiagnosed as SIDS (Shatz et al, 1997).
In our investigation in all tissue samples (except one case), no pathologic findings were found using conventional histologic stainings, especially no signs of myocarditis according to the Dallas criteria. In six cases the detection of enteroviral genome correlated with the immunohistochemical detection of enteroviral capsid protein VP1. Therefore we speculate that enteroviral myocarditis could be regarded as the cause of death in these six cases. Parvovirus B19 could be detected in eight cases of the SIDS group and in three cases of children with well-known causes of death. But all children of the control group were older than 1 year. Because parvovirus B19 infection is common in humans, a final statement concerning myocarditis in cases of suspected SIDS is not possible. We were not able to get reproducible results by immunohistochemical analysis of parvovirus B19 because of the highly degraded autopsy samples.
Although we used sensitive and specific PCR assays for two groups of viruses that together account for a high number of cases of myocarditis in infancy in whom diagnosis is established, it is possible that the myocardial pathology in the PCR-negative infants was caused by infection with viruses other than enteroviruses and parvovirus B19. Further investigations with other viruses, such as adenovirus and Epstein-Barr virus, must be studied. Other possible causes of false-negative PCR assays include uneven distribution of viral genome in the tissues, low titer of virus, or poor preservation of the viral genome in the paraffin-embedded samples. Analysis of additional samples from other regions of the heart might have increased the percent of infants in whom viral genome was detected. Alternatively the myocardial pathology might have resulted from an immune response triggered by a virus that is no longer present in the myocardium at the time of death.
In the present study, the myocardial tissues had been fixed in neutral buffered formalin (pH 7.0) or in NoToX as an alternative fixative (Earth Safe Industries, Inc.) for a maximum of 48 hours. This was crucial for the extraction of intact nucleic acid. Only highly degraded DNA and RNA could be isolated from tissues that had been fixed in formaldehyde for months up to years, as demonstrated by amplification of the housekeeping gene. In those cases reproducible PCR-based virus diagnosis was not possible.
Because of the high potential sensitivity of PCR, precautions must be observed to exclude the possibility of contamination of a reaction tube with previously amplified PCR product or target RNA or DNA from another individual. In this study a number of procedures were adopted to eliminate this risk. The processing of heart samples and preparation of reagents were performed in a room different from that used for PCR amplification and analysis of amplification products. Dedicated reagents, micropipettes, sterile reagent tubes, and filtered pipette tips were used for RNA extraction and RT-PCR assembly. We included negative control blank extractions in which no positive control virus or RNA was added. Positive control samples on each occasion and equal numbers of test samples and reagent blanks were included in each RT-PCR assay.
In conclusion, the ability to isolate intact viral DNA and RNA from paraffin-embedded tissue permits analysis of several viruses that cause myocarditis. Our results emphasize the importance of the PCR-based diagnosis in cases of sudden death even when histologic examination revealed no serious findings in heart muscle tissue.
Materials and Methods
Myocardial tissue from 60 cases after autopsy that were classified as SIDS and 28 cases with known cause of death (children 0–18 years old) had been fixed in neutral buffered formalin (pH 7.0) or in an alternative fixative (NoToX) for a maximum of 48 hours. The tissue was then embedded in paraffin using an automatic system (Shannon Citadel 1000), resulting in a total number of 176 paraffin blocks.
DNA/RNA Extraction
Paraffin-embedded myocardial samples were dewaxed with xylene and washed with ethanol. The paraffin sections were homogenized using a Miccra D-8 homogenizer (Art-moderne Labortechnik, Müllheim-Hügelheim, Germany). Total RNA was extracted with peqGOLD TriFast (peqlab; Biotechnology, Erlangen, Germany) and genomic/viral DNA was extracted with the Gen-ial First-DNA Kit (Gen-ial, Troisdorf, Germany) according to the supplier’s protocol. Contamination was controlled between homogenization of samples as follows: the homogenizer was washed with diethylpyrocarbonate-treated dH2O for 1 minute and then washed with 1% SDS for 1 minute before being placed in 100% ethanol for 1 minute. Because enteroviruses are RNA viruses, RT-PCR was used to evaluate these viruses. First-strand cDNA, for use in detection of enterovirus, was generated from <50 ng of extracted total nucleic acid in the presence of 10 U of RNase inhibitor using Sensiscipt Reverse Transcriptase (Qiagen, Hilden, Germany) following the manufacturer’s protocol.
Enterovirus PCR
For the amplification of enteroviruses, PCR was conducted in two stages as follows: (1) first stage: 5 μl of first-strand cDNA was combined with 10 pmol/μl primer (EV1b, EV2b), 200 μm of each dNTP, 2.5 μl of 10× PCR buffer, 1.5 mm MgCl2, and 1 U of HotStarTaq DNA polymerase (Qiagen, Hilden, Germany); and (2) second stage: 2 μl of the PCR product from the first stage, 10 pmol/μl primer (EV2b, Entero b), 200 μm of each dNTP, 2.5 μl of 10× PCR buffer, 1.5 mm MgCl2, and 1 U of HotStarTaq DNA polymerase (Qiagen). Amplification was performed on a Biometra, Triothermoblock at an initial 15-minute incubation at 94° C. Thirty rounds (for the first PCR) and 20 rounds (for the second PCR) of amplification were performed under the following conditions: 94° C for 30 seconds, 58° C for 30 seconds, and 72° C for 1 minute. The lengths of the amplification products were 150 bp (first round) and 107 bp (second round). Primer sequences were as follows: EV1b, 5′CAATTGTCACCATAAGCAGCCA 3′; EV2b 5′GGCCCCTGAATGCGGCTAAT 3′, Entero b (Martin et al, 1994).
Parvovirus B19 PCR
For the detection of parvoviruses, we used a modified protocol according to the method of Cassinotti et al (1993). Stages were as follows: (1) first stage: 3 μl of extracted total DNA was combined with 10 pmol/μl primer, 200 μm of each dNTP, 2.5 μl of 10× PCR buffer, 1.5 mm MgCl2, and 1 U of HotStarTaq DNA polymerase (Qiagen); and (2) second stage: 2 μl of the PCR product from the first stage, 10 pmol/μl primer, 200 μm of each dNTP, 2.5 μl of 10× PCR buffer, 1.5 mm MgCl2, and 1 U of HotStarTaq DNA polymerase (Qiagen). Amplification was included a 15-minute initial denaturation at 94° C; 30 rounds (for the first PCR) and 20 rounds (for the second PCR) of amplification were performed at the following conditions: 94° C for 30 seconds, 50° C for 30 seconds (first round), and 60° C for 30 seconds (second round), and 72° C for 1 minute. The lengths of the amplification products were 200 bp (first round) and 120 bp (second round). Primer sequences used were according to Cassinotti et al (1993).
Cyclophilin A PCR
Control PCR amplification to verify the presence of amplifiable nucleic acid extract from each sample was performed using cyclophilin a primers. Five microliters of cDNA or 3 μl of DNA was combined with 10 pmol/μl primer, 200 μm of each dNTP, 2.5 μl of 10× PCR buffer, 1.5 mm MgCl2, and 1 U of HotStarTaq DNA polymerase (Qiagen).
Amplification included a 15-minute initial denaturation at 94° C. Thirty-five rounds were performed at the following conditions: 94° C for 30 seconds, 58° C for 30 seconds, and 72° C for 1 minute. The length of the amplification product was 180 bp. Primer sequences were as follows: Cyc F, 5′GTCCAGCAT-TTGCCATGGA-3′; Cyc R, 5′-GACAAGGTCCCAAA-GACAG-3′.
Electrophoresis
Four microliters of each reaction was analyzed on a 8% polyacrylamide gel. Gels were cast on Gelbond (Biozym) and were 0.75-mm thick with 0.07 m Tris sulfate in the gel and 0.28 m Tris borate buffer in the agarose plugs. The electrophoresis was run for 8 cm. The DNA fragments were detected by silver staining.
To avoid false-positive results by contamination, the preventive measures of Kwok and Higuchi (1989) were followed, and negative controls (ie, no template) were included in all experiments. For each assay, known positive controls (infected viral cells) were also added.
The purified PCR products were sequenced directly on an automated ABI model 310 sequencer. Sequence comparison was performed by a BLAST search of GenBank databases.
References
Bendig JWA, O’Brien PS, Muir P, Porter HJ, and Caul EO (2001). Enterovirus sequences resembling coxsackievirus A2 detected in stool and spleen from a girl with fatal myocarditis. J Med Virol 64: 482–486.
Cassinotti P, Weitz M, and Siegl G (1993). Human parvovirus B19: Routine diagnosis by a new nested polymerase chain reaction assay. J Med Virol 40: 228–234.
Cherry JD (1999). Parvovirus infection in children and adults. Adv Pediatr 46: 245–269.
Cohen BJ and Buckley MM (1988). The prevalence of antibody to human parvovirus B19 in England in Wales. J Med Microbiol 25: 151–153.
Grady KL and Costanzo-Nordin MR (1989). Myocarditis: Review of a clinical enigma. Heart Lung 18: 347–355.
Jin O, Sole M, Butany JW, Chia WK, McLaughlin PR, Liu P, and Liew CC (1990). Detection of enterovirus RNA in myocardial biopsies from patients with myocarditis and cardiomyopathy using gene amplification by polymerase chain reaction. Circulation 82: 8–16.
Kwok S and Higuchi R (1989). Avoiding false positives with PCR. Nature 339: 237–238.
Lau G (1994). Acute fulminant, fatal coxsackie B virus infection: A report of two cases. Ann Acad Med Singapore 23: 917–920.
Lozinski GM, Davis GG, Krous HF, Billman GF, Shimizu H, and Burns JC (1994). Adenovirus myocarditis: Retrospective diagnosis by gene amplification from formalin-fixed, paraffin-embedded tissues. Hum Pathol 25: 831–834.
Martin AB, Webber S, Fricker FJ, Jaffe R, Demmler G, Kearney D, Zhang YH, Bodurtha J, Gelb B, Ni J, Bricker JT, and Towbin JA (1994). Acute myocarditis: Rapid diagnosis by PCR in children. Circulation 90: 330–339.
Murry CE, Jerome KR, and Reichenbach DD (2001). Fatal parvovirus myocarditis in a 5-year-old girl. Hum Pathol 32: 342–345.
Rambaud C, Cieuta C, Canioni D, Rouzioux C, Lavaud J, Hubert P, Brousse N, Rudler M, and Cheron G (1992). Cot death and myocarditis. Cardiol Young 2: 266–272.
Shatz A, Hiss J, and Arensburg B (1997). Myocarditis misdiagnosed as sudden infant death syndrome (SIDS). Med Sci Law 37: 16–18.
Towbin JA, Ni J, Demmler G, Martin A, Kearney D, and Bricker JT (1993). Evidence for adenovirus as common cause of myocarditis in children using polymerase chain reaction (PCR). Pediatr Res 33: 27A.
Acknowledgements
The authors thank Ms. Renate Klemmer, Ms. Helga Körner, and Ms. Claudia Schäfer for excellent technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft (DE 814) and the Förderverein Rechtsmedizin der Universität Bonn e.V.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baasner, A., Dettmeyer, R., Graebe, M. et al. PCR-Based Diagnosis of Enterovirus and Parvovirus B19 in Paraffin-Embedded Heart Tissue of Children with Suspected Sudden Infant Death Syndrome. Lab Invest 83, 1451–1455 (2003). https://doi.org/10.1097/01.LAB.0000092232.51370.66
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000092232.51370.66
This article is cited by
-
Letale lymphozytäre Myokarditis – eine unterschätzte Diagnose im Säuglings- und Kindesalter?
Die Pathologie (2023)
-
Forensische Molekularpathologie
Rechtsmedizin (2014)
-
Lethal influenza B myocarditis in a child and review of the literature for pediatric age groups
Infection (2010)
-
Virusinduzierte Myokarditis beim mutmaßlichen plötzlichen Kindstod
Rechtsmedizin (2008)
