
Abstract
We describe a novel autoimmune disease characterized by severe subepidermal bullous eruption and crescentic glomerulonephritis with autoantibodies directed against the noncollagenous domain of the α5 and α6 chains of type IV collagen. Biopsy of perilesional skin revealed a subepidermal blister with marked polymorphonuclear infiltrate with linear deposits of IgA and C3. Light microscopy of a kidney biopsy specimen revealed a crescentic glomerulonephritis, and immunofluorescence microscopy showed linear basement membrane staining for IgA (3+), C3 (1+), and IgG (1+). No electron-dense deposits were observed by transmission electron microscopy. The patient's autoantibodies reacted with normal human skin and kidney: IgA (3+) and IgG (1+) antibodies stained the basement membrane zones of skin, renal glomerulus, and some tubules. The identity of the target antigen was determined by immunochemical analyses of candidate antigens using the patient's autoantibodies. The patient's IgA and IgG autoantibodies reacted with a 185- to 190-kDa antigen from a human dermal extract that was distinguished from the other dermal or epidermal antigens, including the 145- to 290-kDa (type VII collagen) epidermolysis bullosa acquisita antigen, the 165- to 200-kDa α3 laminin mucous membrane cicatricial pemphigoid antigen, and the 230-kDa and the 180-kDa bullous pemphigoid antigens. Patient's IgA and IgG autoantibodies further reacted with the α5(IV) and weakly with the α6(IV) chains of type IV collagen by Western blot and ELISA. This report expands the repertoire of bullous skin disorders and provides an explanation for the association of anti-type IV collagen autoantibodies and glomerulonephritis with subepidermal blisters.
Similar content being viewed by others


Introduction
Type IV collagen is a heterotrimeric complex of proteins composed of α1(IV) through α6(IV) subunit polypeptides (Ghohestani et al, 2001a; Prockop and Kivirikko, 1995). The noncollagenous (NC1) domain of α3(IV) has been identified as the target of an autoimmune response in Goodpasture's syndrome (GP) (Saus et al, 1988). Recently, the NC1 domain of α5(IV) has been characterized as the target of IgG autoantibodies in a novel autoimmune disease characterized by glomerulonephritis and subepidermal blisters (Ghohestani et al, 2000). We now present a novel case with both skin and kidney involvement with the predominant IgA class autoantibodies to the α5(IV) and α6(IV) chains of type IV collagen. Accordingly, the presence of antibodies to the α5(IV) and α6(IV) chains of type IV collagen should be searched in sera from patients who present with subepidermal blisters and glomerulonephritis.
Results
Case Presentation
Serum, skin, and kidney samples were obtained from a 72-year-old white male who presented in January 1998 with an 8-month history of recurrent, diffuse, pruritic, sometimes vesicular eruptions and a 3-week history of submandibular and cervical lymphadenopathy. By history, the lesions improved with systemic steroids, but recurred within several months; the interval between recurrences became shorter with each episode. Past medical history included hypertension and a pacemaker insertion in 1995. There was no history of gross hematuria, arthralgia, or hemoptysis. However, a urinalysis in September of 1997, when the patient had skin lesions, showed 2+ hematuria but no proteinuria. His medications included pindolol, hydrochlorothiazide, and aspirin. Physical examination revealed ill-defined erythematous macules and patches in addition to eroded papules on his anterior and posterior trunk and bilaterally on the upper and lower extremities. Additionally, he had an erosion on his left upper lip.
Biopsy of perilesional skin revealed a subepidermal blister with marked polymorphonuclear (PMN) infiltrate. Direct immunofluorescence (IF) of skin samples revealed linear deposits of IgA (3+) and C3 (1+) along the dermal-epidermal junction. The patient's history and physical and immunopathologic examinations were all suggestive of linear IgA bullous dermatosis (LABD), and he was placed on doxycycline (100 mg tid). In December 1999, the patient presented with erosions bilaterally on the upper and lower extremities and the tongue associated with symblepharon in both eyes. A prednisone taper beginning at 60 mg/day and azathioprine (100 mg daily) were instituted. In January 2000, the patient had new skin lesions on his hands and knees as well as significant ulcerations in his mouth, predominantly on his tongue and palate. Skin lesions healed without significant scarring. Extensive milia formation was, however, noted in healed areas. Medications included amlodipine (10 mg po qd), doxycycline (100 mg po tid), prednisone (20 mg po qd), and citalopram hydrobromide (20 mg po qd).
Laboratory studies revealed the following: serum creatinine 5.6 mg/dl (495 μmol/L), blood urea nitrogen 122 mg/dl (43.5 mmol/L), glucose 108 mg/dl (6 mmol/L), sodium 140 mEq/L (140 mmol/L), potassium 4.3 mEq/L (4.3 mmol/dl), chloride 104 mEq/L (104 mmol/L), bicarbonate 22 mEq/L (22 mmol/L), hemoglobin 8.0 g/dl (80 g/L), hematocrit 23.2%, platelets 213 × 109/L, white blood cell count 11.0 × 109/L, albumin 2.9 g/dl (29 g/L), and cholesterol 158 mg/dl (4.1 mmol/L). The urine dipstick revealed 3+ protein and 3+ blood, and microscopic examination of the sediment showed many red blood cells and moderate granular casts. The urine protein:creatinine ratio was 6.2. Renal ultrasound showed normal-sized kidneys without hydronephrosis. The following were either normal or negative: antinuclear antibody, hepatitis C antibody, hepatitis B surface antigen, haptoglobin, C3, C4, serum IgA, and antineutrophil cytoplasmic antibodies. An ultrasound-guided left percutaneous renal biopsy was performed. Light microscopy revealed a crescentic glomerulonephritis (Fig. 1A). The core needle biopsy contained eight glomeruli, four with crescents and two globally sclerotic. Two nonsclerotic, crescentic glomeruli exhibited periglomerular fibrosis and were hypercellular with an increase in mesangial matrix and cells. The capillary loops were collapsed and in many cases obliterated. Trichrome stain revealed the presence of collagen in these same glomeruli, and silver stain showed reduplication of the glomerular basement membranes (GBM). Tubules were atrophic, and there was a moderate chronic interstitial inflammatory infiltrate. Intralobular arteries showed fibrous intimal thickening. IF microscopy showed linear basement membrane staining for C3 (1+), IgA (3+), and IgG (1+). No electron-dense deposits were observed by transmission electron microscopy (Fig. 1B). The patient was treated with pulse methylprednisolone (1000 mg iv qd × 3 days) followed by oral prednisone (40 mg po qd), cyclophosphamide (150 mg po qd), and plasmapheresis. The patient's skin lesions dramatically improved after treatment. However, his course was complicated by catheter-related Staphylococcus aureus bacteremia. Despite aggressive therapy, the serum creatinine level rose to 8.6 mg/dl in March 2000. The patient began peritoneal dialysis in August 2000. Historic serum creatinine values are demonstrated in Figure 2.

Crescentic glomerulonephritis in the absence of electron-dense deposits is suggestive of an anti-glomerular basement membrane (GBM) pathology. A, Light microscopy of kidney section shows sheets of proliferating epithelial cells that fill Bowman's space. The glomerular tufts are collapsed and exhibit mesangial cell proliferation (magnification, × 400). B, Transmission electron microscopy reveals epithelial cells that have fused foot processes with peripheral condensation of actin. There are no electron-dense deposits. The bar represents 500 nm.

Comparison between serum creatinine level and severity of skin lesions. Onset of the disease is associated with almost doubling the serum creatinine level to 1.9 mg/dL. Severe blister formation in January 2000 is also associated with a significant rise in serum creatinine to 5.6 mg/dL. Severity of the skin involvement is classified as none (0), mild (1), moderate (2), severe (3), and very severe (4), based on the number of individual lesions (see Materials and Methods).
Characterization of the Autoantibodies
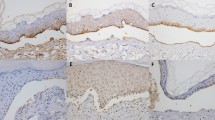
Indirect IF of the patient's serum revealed binding of IgA (3+) and IgG (1+) antibodies along the dermal-epidermal junction (Fig. 3A), specifically on the dermal side of 1.0 M NaCl-split skin (Fig. 3B). On normal human kidney sections, the patient's circulating IgA (3+) (Fig. 3C), and to a lesser extent IgG (1+), reacted with the GBM. This reactivity was in contrast to LABD, epidermolysis bullosa acquisita (EBA), and bullous pemphigoid (BP) autoantibodies that fail to react with the GBM (Fig. 3D).

Immunolocalization of antigen recognized by the patient's autoantibodies by immunofluorescence (IF) microscopy on frozen skin and kidney sections. A, IF microscopy of the patient's serum revealed the predominant presence of IgA antibodies along the dermal-epidermal junction. B, IgA antibodies were primarily directed against a component of skin basement membrane zone, which lies on the dermal side of 1.0 M NaCl-split skin. The level of the split was checked to be within the lamina lucida in 1.0 M NaCl-split skin; antibodies to α6β4 integrins labeled the epidermal side and anti-laminin-5 antibodies the dermal side of salt-split skin. C, The patient's IgA antibodies clearly reacted with the GBM of a normal kidney section; tubular basement membrane was weakly stained. D, An epidermolysis bullosa acquisita (EBA) serum, used as a control, failed to react with the GBM. Arrows indicate the specific IgA deposits. E = epidermis; D = dermis.
The identity of the target antigen was determined by immunochemical analyses of candidate antigens using the patient's serum samples. The patient's IgA and IgG autoantibodies reacted with a 185- to 190-kDa antigen from a human dermal extract (Fig. 4A), different from the other dermal antigens, including the 145- to 290-kDa (type VII collagen) EBA antigen (Fig. 4A) and the 165- to 200-kDa α3 chain of laminin (cicatricial pemphigoid [CP] antigen) (Fig. 4B). The patient's serum samples did not react with the 230-kDa and the 180-kDa BP antigens. IgA and IgG antibodies eluted from the 185- to 190-kDa protein, fractionated by electrophoresis, reacted with the skin and renal basement membrane zone (BMZ) in an analogous fashion with the whole serum (data not shown). The patient's IgA autoantibodies further reacted with the α5(IV) and weakly with the α6(IV) chains of type IV collagen by Western blot (Fig. 4C). IgG autoantibodies from the patient serum also reacted weakly with the α5(IV) and α6(IV) chains by Western blot (data not shown).

Characterization of the target antigen by Western blot using anti-IgA (α chain specific) or anti-IgG (γ chain specific) conjugates on various substrates. A, Dermal protein extracts. The patient's IgA labeled a 185- to 190-kDa molecule (lane 1) that comigrated with the α5(IV) collagen chain as labeled by a specific antibody (lane 3); type VII collagen was labeled by serum from a patient with EBA (lane 2); none of the proteins in the extract were labeled by serum from a healthy individual (lane 4). B, Purified laminin-5. The patient's IgA serum (lane 1) did not label the 165- to 200-kDa α3 laminin identified by a rabbit polyclonal antibody (lane 2). C, Recombinant α5(IV) noncollagenous (NC1) domain. The patient's serum IgA labeled the α5(IV) NC1 domain at 1:100 dilution (lane 1). The 25-kDa recombinant α5(IV) NC1 domain that is bound to a Flag sequence was labeled by an anti-Flag antibody (lane 3) but not by a normal human serum (lane 2). Proteins were separated by SDS-PAGE using a 4% acrylamide gel for dermal lysates, gradient 4% to 20% for purified laminin-5, and 10% for recombinant proteins. The patient's serum is indicated by an arrow.
By ELISA, the patient's serum IgA reacted with the recombinant NC1 domain of the α5(IV) (OD 0.58) and the α6(IV) (OD 0.39), among the six NC1 domains of type IV collagen (Fig. 5). The patient's serum IgG reacted weakly with the α5(IV) (OD 0.29) and the α6(IV) (OD 0.2). An anti-α5(IV) human serum reacted strongly with the α5(IV) chain (OD 0.71) by ELISA. Other sera tested, either obtained from healthy individuals or from patients with LABD, EBA, BP, or CP, did not react with the α1(IV) through α6(IV) chains of type IV collagen by ELISA (OD below 0.14) (Fig. 5).

ELISA reactivity of the patient's serum with recombinant proteins corresponding to the six NC1 domains of type IV collagen chains (α1–α6) using anti-IgA and anti-IgG conjugates. The patient's serum contained predominantly IgA antibodies to collagen α5(IV) and α6(IV) NC1 recombinant proteins. IgG autoantibodies to α5(IV) and α6(IV) NC1 were also present but at a lower level. The positive control serum from a patient with IgG anti-α5(IV) strongly reacted with the α5(IV) NC1. Sera from a healthy individual and from a patient with EBA with antibodies to type VII collagen remained nonreactive with all six NC1 domains. Cut-off values for positive samples were set at A = 0·14 (dotted line).
Discussion
We present a novel case with both skin and kidney involvement with the predominant IgA class autoantibodies to the α5(IV) and α6(IV) chains of type IV collagen. This case is distinguished from several immune-mediated diseases including lupus erythematosus, GP, IgA nephropathy, LABD, EBA, BP, CP, and pemphigus. It differs from GP because of the antigenic specificity of the autoantibodies (Salama et al, 2001). IgG autoantibodies in GP are directed against the NC1 domain of the α3(IV) collagen chain (Saus et al, 1988). Lack of mesangial deposits as determined by IF and transmission electron microscopy ruled out IgA nephropathy (Couser, 1999). Absence of arthritis and neurologic and hematologic disorders, as well as lack of subendothelial and mesangial deposits on the kidney biopsy specimen, were all strongly suggestive against lupus erythematosus (Petri, 1998). This case also differs from other blistering skin diseases because of the kidney involvement and antigenic specificity of the patient's autoantibodies. In LABD, the autoantibodies are predominantly of the IgA class, bind the epidermal side of salt-split skin, and are directed against the whole (180 and 230 kDa) or degradation fragments of BP antigens (97 and 120 kDa) (Ghohestani et al, 1997a; Zone et al, 1998). These antigens are not expressed, however, along the GBM (Ghohestani et al, 2000). In BP, characterized by subepidermal blisters, IgG autoantibodies are directed against the 180- and 230-kDa BP antigens (Ghohestani et al, 2001b; Stern, 2002; Yancey and Egan, 2000). CP rarely exhibits widespread skin lesions, and if observed, they are accompanied by scarring; the antibodies in CP are targeted to the 180-kDa BP antigen, laminin-5, or an uncharacterized 168-kDa molecule (Cotell et al, 2000. In EBA, autoantibodies target the type VII collagen and are primarily of IgG subclass (Hallel-Halevy et al, 2001). In pemphigus, antibodies are directed against the desmosomal antigens and induce intraepidermal blisters (Anhalt and Diaz, 2001; Edelson, 2000).
By IF microscopy, the patient's IgA and IgG autoantibodies reacted with the dermal side of salt-split skin, displaying similar patterns seen with sera from patients with EBA and anti-laminin-5 CP (Ghohestani et al, 1997b); but in contrast to those sera, the autoantibodies in our case stained the GBM. Anti-GBM reactivity of the patient's autoantibodies accounts for the development of glomerulonephritis as documented by light, IF, and electron microscopy of the kidney biopsy specimen.
The identity of the target antigen was established by biochemical analyses of several candidate antigens. The epithelial basement membrane is a complex structure with more than 20 identified gene products including type IV collagen (α1, α2, α5, and α6 chains), the 230-kDa and the 180-kDa (type XVII collagen) BP antigens, type VII collagen, laminin-5, and the α6β4 integrin (Ghohestani et al, 2001a). Some of these components, such as the 230-kDa (Mueller et al, 1989) and the 180-kDa BP antigens (Diaz et al, 1990), type VII collagen (Woodley et al, 1988), laminin-5 (Egan and Yancey 2000; Ghohestani et al, 1997b), and β4 integrin (Bhol et al, 1996), have been identified and characterized as the target in different forms of blistering skin diseases. Recently, collagen α5(IV) has been added to this list as the target of autoantibodies in patients presenting with skin blisters and glomerulonephritis (Ghohestani et al, 2000). Our present case showed similar clinical features characterized by the presence of subepidermal blisters and glomerulonephritis. However, the autoantibodies in our case are predominantly of the IgA class and they are directed against both the α5(IV) and α6(IV) collagen chains. No IgA or IgG reactivity was observed against other components of the epithelial basement membrane. These findings suggest the presence of a novel subtype of anti-type IV collagen autoimmune disease with predominant involvement of IgA autoantibodies. It should be noted that a variant of GP, characterized by the presence of IgA anti-GBM autoantibodies, has been described (Lee and Marks, 1999). Autoantibodies in these patients were, however, directed against the collagen α3(IV) chain without any skin involvement (Fervenza et al, 1999).
Type IV collagen molecules form a network structure primarily in the basement membranes of various tissues. To date, six genetically distinct type IV collagen polypeptide chains, α1(IV) through α6(IV), have been described (Hudson et al, 1993). The α1(IV) and α2(IV) chains, encoded by the COL4A1 and COL4A2 genes, respectively, are ubiquitous, whereas the α3(IV), α4(IV), α5(IV), and α6(IV) chains, encoded by the COL4A3, COL4A4, COL4A5, and COL4A6 genes, respectively, are present in a restricted tissue distribution. The α5(IV) and α6(IV) chains are both present at the dermal-epidermal junction, whereas the α3(IV), α4(IV), and α5(IV) chains are expressed along the GBM (Boutaud et al, 2000; Ghohestani et al, 2000).
The erythematous base of the cutaneous lesions and the marked PMN cell infiltrates observed in the perilesional biopsy specimen from our case suggested an important role for PMNs in skin BMZ disruption, as has already been demonstrated in BP (Delaporte et al, 1996; Liu et al, 1997). PMNs could play a crucial role in the dermal-epidermal separation through the release of their enzymatic mediators (Delaporte et al, 1996). Accordingly, this could explain the absence of major skin lesions in X-linked Alport syndrome, with mutations in COL4A5 (Dagher et al, 2001). The features of Alport syndrome reflect derangement of basement membrane structure and function resulting from changes in type IV collagen expression (Kashtan, 1999). The primary pathologic event seems to be the loss of a type IV collagen network composed of the α3(IV), α4(IV), and α5(IV) chains from the GBM. Although this network is not critical for normal glomerulogenesis, its absence seems to provoke the overexpression of other extracellular matrix proteins, such as the α1(IV) and α2(IV) collagen chains leading to glomerulosclerosis (Kashtan, 1999). In skin, the absence of α5(IV) and α6(IV) chains could be largely compensated by the classic type IV collagen molecule, [α1(IV)]2α2(IV), stabilizing the skin BMZ, whereas the presence of autoantibodies against these proteins could lead to disruption of skin BMZ mainly through complement fixation and recruitment of inflammation cell infiltrates, followed by release and activation of proteases, as has been shown in a mouse model of BP (Liu et al, 1997).
Antibodies to the α5(IV) chain have been also found in patients with Alport syndrome who received a normal kidney that expresses the wild type of the α5(IV) chain (Brainwood et al, 1998). Their skin BMZ does not, however, express α5(IV); consequently, no blistering can be seen in these patients. Development of alloantibodies in these cases further attests to the immunogenicity of the collagen α5(IV) chain in humans.
Association of subepidermal blisters and glomerulopathy has been previously described (Barnadas et al, 1998; Ross and Ahmed, 1989; van Joost et al, 1986). Most cases had IgA or IgG deposits in both the skin and glomerular BMZ (Davenport et al, 1987). However, the antigenic specificity of the patients' autoantibodies was not determined. Some patients were also diagnosed as having an extension of GP with skin involvement (Davenport et al, 1987). However, the skin BMZ is devoid of α3(IV), the GP target antigen (Ghohestani et al, 2000). Collectively, the present data demonstrate that anti-GBM disease can be associated with skin blisters caused by circulating antibodies recognizing type IV collagen antigenic epitopes. Accordingly, the presence of antibodies to type IV collagen chains should be examined in patients who present with subepidermal blisters and glomerulonephritis.
Materials and Methods
Control sera were obtained from the following: (i) two patients with GP with IgG autoantibodies directed against the α3 chain of type IV collagen; (ii) one patient with antibodies to the α5 chain of type IV collagen (Ghohestani et al, 2000); (iii) two patients with EBA with circulating antibodies to the 290-kDa antigen representing type VII collagen; (iv) two BP patients with IgG antibodies to the 180-kDa and the 230-kDa BP antigens 1 and 2, respectively; (v) two patients with LABD with IgA antibodies to the 97- to 180-kDa antigen; (vi) two CP patients with circulating antibodies to the 165- to 200-kDa α3 chain of laminin-5; and (vii) eight unrelated healthy individuals.
The monoclonal and polyclonal antibodies used in this study were as follows: (i) a rabbit polyclonal antibody to type IV collagen purified from human placenta (Institut Pasteur de Lyon, Lyon, France); (ii) an mAb to either the α1(IV) or α3(IV) chain of type IV collagen (Wieslab, Lund, Sweden); (iii) polyclonal antibodies specific for the α5(IV) and α6(IV) chains of type IV collagen raised in mice in our laboratory (Dr. Ghohestani); (iv) a mouse mAb to type IV collagen (Dako SA, Trappes, France); (v) a mouse mAb to type VII collagen (Serotec, Oxford, England); (vi) a human mAb to the 230-kDa BP antigen (BP230) (Peyron et al, 1994); (vii) a rabbit polyclonal antibody to the immunodominant epitopes of the 180-kDa (BP180) ectodomain (R67) (Ghohestani et al, 1988); and (viii) a rabbit antibody to laminin-5 produced in our laboratory (Dr. Ghohestani).
Circulating anti-BMZ antibodies were detected by IF performed on 4-μm cryostat sections of the following normal human tissue substrates: skin, salt-split skin, and kidney. In salt-split skin, the dermal-epidermal junction was separated by 1 M NaCl containing 50 μm phenylmethylsulphonyl fluoride and 0.1 M EDTA (Ghohestani et al, 1988, 1996). Intensity of positive reactivity by IF microscopy was rated as follows: 3+, strong; 2+, moderate; and 1+, weak. Severity of skin lesions was rated as follows: 4+, very severe, if number of blisters (x) was >41; 3+, severe, if 40 ≥ x > 21; 2+, moderate, if 20 ≥ x > 6; 1+, mild, if 5 ≥ x > 0; and free of lesion if x = 0. This classification was made on the basis of frequency of distribution for number of blisters.
To evaluate the immunoreactivity of the patient's serum against the type IV collagen chains, we produced fusion proteins corresponding to the NC1 domains of the α5(IV) and α6(IV) chains (Ghohestani et al, 2000). The secreted proteins carried the Flag sequence fused to the amino terminus of the full-length NC1 domains. The proteins were purified by a single-affinity chromatographic step using anti-Flag agarose columns.
To rule out the presence of autoantibodies to skin antigens other than type IV collagen chains, we tested the patient's serum against dermal and epidermal protein lysates derived from human dermis and cultured human keratinocytes, respectively (Ghohestani et al, 2000). Furthermore, the patient's serum samples were tested against the recombinant proteins coding for the 230-kDa and the 180-kDa (type XVII collagen) BP antigens as well as the native purified laminin-5 and type VII collagen by Western blot (Ghohestani et al, 2000; Rousselle et al, 1997). Purified native or recombinant proteins were separated by SDS-PAGE (4–22% acrylamide gels) under reducing conditions, transferred onto nitrocellulose membranes, and incubated with the sera at 1:100 dilution, as described elsewhere (Ghohestani et al, 1996). Antibodies recognizing the protein of interest (185–190 kDa), blotted onto nitrocellulose membrane, were affinity purified (Ghohestani et al, 2000). ELISA reactivity of antibodies was performed on plates coated with 1 μg/well purified recombinant collagen α1(IV) through α6(IV) chains, as described (Ghohestani et al, 2000). A cutoff value was defined as the average value +3 sd of negative control sera. Each sample was run in duplicate.
References
Anhalt GJ and Diaz LA (2001). Prospects for autoimmune disease: Research advances in pemphigus. JAMA 285: 652–654.
Barnadas MA, Gelpi C, Rocamora V, Baro E, Ballarin J, Nadal C, Bielsa A, Arostegui J, and Alomar A (1998). Bullous pemphigoid associated with acute glomerulonephritis. Br J Dermatol 138: 867–871.
Bhol KC, Dans MJ, Simmons RK, Foster CS, Giancotti FG, and Ahmed AR (1996). The autoantibodies to alpha 6 beta 4 integrin of patients affected by ocular cicatricial pemphigoid recognize predominantly epitopes within the large cytoplasmic domain of human beta 4. Proc Natl Acad Sci USA 93: 14714–14719.
Boutaud A, Borza DB, Bondar O, Gunwar S, Netzer KO, Singh N, Ninomiya Y, Sado Y, Noelken ME, and Hudson BG (2000). Type IV collagen of the glomerular basement membrane: Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J Biol Chem 275: 30716–30724.
Brainwood D, Kashtan C, Gubler MC, and Turner AN (1998). Targets of alloantibodies in Alport anti-glomerular basement membrane disease after renal transplantation. Kidney Int 53: 762–766.
Cotell S, Robinson ND, and Chan LS (2000). Autoimmune blistering skin diseases. Am J Emerg Med 18: 288–299.
Couser WG (1999). Glomerulonephritis. Lancet 353: 1509–1515.
Dagher H, Buzza M, Colville D, Jones C, Powell H, Fassett R, Wilson D, Agar J, and Savige J (2001). A comparison of the clinical, histopathologic, and ultrastructural phenotypes in carriers of X-linked and autosomal recessive Alport's syndrome. Am J Kidney Dis 38: 1217–1228.
Davenport A, Verbov JL, and Goldsmith HJ (1987). Circulating anti-skin basement membrane zone antibodies in a patient with Goodpasture's syndrome. Br J Dermatol 117: 125–127.
Delaporte E, Dubost-Brama A, Ghohestani R, Nicolas JF, Neyrinck JL, Bergoend H, Janin A, and Capron M (1996). IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol 157: 3642–3647.
Diaz LA, Ratrie H 3rd, Saunders WS, Futamura S, Squiquera HL, Anhalt GJ, and Giudice GJ (1990). Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera: Immunolocalization of this protein to the hemidesmosome. J Clin Invest 86: 1088–1094.
Edelson RL (2000). Pemphigus: Decoding the cellular language of cutaneous autoimmunity. N Engl J Med 343: 60–61.
Egan CA and Yancey KB (2000). The clinical and immunopathological manifestations of anti-epiligrin cicatricial pemphigoid, a recently defined subepithelial autoimmune blistering disease. Eur J Dermatol 10: 585–589.
Fervenza FC, Terreros D, Boutaud A, Hudson BG, Williams RA Jr, Donadio JV Jr, and Schwab TR (1999). Recurrent Goodpasture's disease due to a monoclonal IgA-kappa circulating antibody. Am J Kidney Dis 34: 549–555.
Ghohestani R, Kanitakis J, Nicolas JF, Cozzani E, and Claudy A (1996). Comparative sensitivity of indirect immunofluorescence to immunoblot assay for the detection of circulating antibodies to bullous pemphigoid antigens 1 and 2. Br J Dermatol 135: 74–79.
Ghohestani RF, Cozzani E, Delaporte E, Nicolas JF, Parodi A, and Claudy A (1988). IgE antibodies in sera from patients with bullous pemphigoid are autoantibodies preferentially directed against the 230-kDa epidermal antigen (BP230). J Clin Immunol 18: 202–209.
Ghohestani RF, Hudson BG, Claudy A, and Uitto J (2000). The alpha 5 chain of type IV collagen is the target of IgG autoantibodies in a novel autoimmune disease with subepidermal blisters and renal insufficiency. J Biol Chem 275: 16002–16006.
Ghohestani RF, Li K, Rousselle P, and Uitto J (2001a). Molecular organization of the cutaneous basement membrane zone. Clin Dermatol 19: 551–562.
Ghohestani RF, Nicolas JF, Kanitakis J, and Claudy A (1997a). Linear IgA bullous dermatosis with IgA antibodies exclusively directed against the 180- or 230-kDa epidermal antigens. J Invest Dermatol 108: 854–858.
Ghohestani RF, Nicolas JF, Rousselle P, and Claudy AL (1997b). Diagnostic value of indirect immunofluorescence on sodium chloride-split skin in differential diagnosis of subepidermal autoimmune bullous dermatoses. Arch Dermatol 133: 1102–1107.
Ghohestani RF, Novotney J, Chaudhary M, and Agah RS (2001b). Bullous pemphigoid: From the bedside to the research laboratory. Clin Dermatol 19: 690–696.
Hallel-Halevy D, Nadelman C, Chen M, and Woodley DT (2001). Epidermolysis bullosa acquisita: Update and review. Clin Dermatol 19: 712–718.
Hudson BG, Reeders ST, and Tryggvason K (1993). Type IV collagen: Structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem 268: 26033–26036.
Kashtan CE (1999). Alport syndrome: An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore) 78: 338–360.
Lee SM and Marks EA (1999). The emerging spectrum of IgA-mediated renal diseases: Is there an IgA variant of Goodpasture's syndrome? Am J Kidney Dis 34: 565–568.
Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, Till GO, and Diaz LA (1997). A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest 100: 1256–1263.
Mueller S, Klaus-Kovtun V, and Stanley JR (1989). A 230-kD basic protein is the major bullous pemphigoid antigen. J Invest Dermatol 92: 33–38.
Petri M (1998). Treatment of systemic lupus erythematosus: An update. Am Fam Physician 57: 2753–2760.
Peyron E, Nicolas JF, Reano A, Roche P, Thivolet J, Haftek M, Schmitt D, Peronne C, Banchereau J, and Rousset F (1994). Human monoclonal autoantibodies specific for the bullous pemphigoid antigen 1 (BPAg 1). J Immunol 153: 1333–1339.
Prockop DJ and Kivirikko KI (1995). Collagens: Molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 64: 403–434.
Ross EA and Ahmed AR (1989). Bullous pemphigoid-associated nephropathy: Report of two cases and review of the literature. Am J Kidney Dis 14: 225–229.
Rousselle P, Keene DR, Ruggiero F, Champliaud MF, Rest M, and Burgeson RE (1997). Laminin 5 binds the NC-1 domain of type VII collagen. J Cell Biol 138: 719–728.
Salama AD, Levy JB, Lightstone L, and Pusey CD (2001). Goodpasture's disease. Lancet 358: 917–920.
Saus J, Wieslander J, Langeveld JP, Quinones S, and Hudson BG (1988). Identification of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV. J Biol Chem 263: 13374–13380.
Stern RS (2002). Bullous pemphigoid therapy: Think globally, act locally. N Engl J Med 346: 364–367.
van Joost T, Muntendam J, Heule F, Stolz E, Vuzevski VD, and Kate F (1986). Subepidermal bullous autoimmune disease associated with immune nephritis: Immunomorphologic studies. J Am Acad Dermatol 14: 214–220.
Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, and Briggaman RA (1988). Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest 81: 683–687.
Yancey KB and Egan CA (2000). Pemphigoid: Clinical, histologic, immunopathologic, and therapeutic considerations. JAMA 284: 350–356.
Zone JJ, Taylor TB, Meyer LJ, and Petersen MJ (1998). The 97 kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180 kDa bullous pemphigoid antigen, BPAg2. J Invest Dermatol 110: 207–210.
Acknowledgements
This work was supported by grants from the National Institutes of Health and the American Skin Association.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ghohestani, R., Rotunda, S., Hudson, B. et al. Crescentic Glomerulonephritis and Subepidermal Blisters with Autoantibodies to α5 and α6 Chains of Type IV Collagen. Lab Invest 83, 605–611 (2003). https://doi.org/10.1097/01.LAB.0000067497.86646.4D
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000067497.86646.4D
This article is cited by
-
An unusual case of IgA-mediated anti-glomerular basement membrane disease
International Urology and Nephrology (2013)
-
Advances in human antiglomerular basement membrane disease
Nature Reviews Nephrology (2011)
