
Abstract
The exact nature of the desmoplastic small round cell tumor (DSRCT) remains controversial. More detailed analyses might be facilitated by the establishment of permanent DSRCT cell lines. To date, however, no human DSRCT cell line has been reported. In this study, we report the establishment of a new human cell line, JN-DSRCT-1, from the pleural effusion of a 7-year-old boy with pulmonary metastasis from a typical intra-abdominal DSRCT. JN-DSRCT-1 cells were small round or spindle shaped with oval nuclei and have been maintained continuously in vitro for over 190 passages during more than 40 months. Histologic features of the heterotransplanted tumors in severe combined immunodeficiency mouse were essentially the same as those of the original DSRCT, revealing nests or clusters of small round cells embedded in an abundant desmoplastic stroma. Both in vitro and in vivo, the cells exhibited immunopositive reactions for vimentin, desmin, cytokeratins (AE1/AE3 and CAM 5.2), epithelial membrane antigen, neuron-specific antigen, and CD57 (Leu-7). JN-DSRCT-1 cells exhibited a pathognomonic t(11;22)(p13;q12) translocation by cytogenetic analysis. In addition, RT-PCR and sequencing analysis revealed a chimeric transcriptional message of the Ewing’s sarcoma gene exon 10 fused to the Wilms’ tumor gene exon 8. To our knowledge, this is the first permanent human DSRCT cell line. The JN-DSRCT-1 cell line, which exhibits the unique morphologic and genetic characteristics of DSRCT, will be extremely useful for a variety of important studies such as the pathogenic mechanism, biologic behavior, and therapeutic model of human DSRCT.
Similar content being viewed by others

Introduction
The desmoplastic small round cell tumor (DSRCT) is a relatively rare malignant tumor that typically involves the abdominal and/or pelvic peritoneum of children and young adults (Weiss and Goldblum, 2001). Despite a distinct clinicopathologic entity, the exact nature of this tumor is not known. Although several authors have suggested a histogenetic relationship to the mesothelium or submesothelial mesenchyme (Gerald et al, 1991; Parkash et al, 1995), such hypothesis often fails to account for tumors located in the brain, hand, parotid, and neck (Adsay et al, 1999; Mihok and Cha, 2001; Tison et al, 1996; Wolf et al, 1999). Recent cytogenetic and molecular studies have indicated that the t(11;22)(p13;q12) translocation and the resulting Ewing’s sarcoma gene-Wilms’ tumor gene (EWS-WT1) fusion gene induced by this translocation are pathognomonic for DSRCT (Argatoff et al, 1996; Biegel et al, 1993; Brodie et al, 1995; de Alava et al, 1995; Gerald et al, 1995; Ladanyi and Gerald, 1994; Rodriguez et al, 1993; Sawyer et al, 1992).
A stable human DSRCT cell line that retains the unique in vitro and in vivo features of DSRCT would facilitate the study of this tumor. To our knowledge, no human cell line derived from DSRCT has been developed. In this report, we describe the development of a novel human cell line, JN-DSRCT-1, derived from an intra-abdominal DSRCT, which may provide a useful in vitro model for the study of the pathogenesis of DSRCT.
Results
Establishment of the JN-DSRCT-1 Cell Line
With the establishment of the primary culture, tumor cells showed excellent growth without contact inhibition from the beginning. Three weeks after initial cultivation in the primary culture, small round or spindle-shaped tumor cells reached subconfluence as well as the appearance of some piled-up cell foci. The cells were collected after a 5-minute digestion at 37° C with a 0.1% trypsin solution and replated in two 25-cm2 plastic flasks containing fresh medium. Thereafter, the cells were serially subcultured at a dilution of 1:2 when confluent. Approximately 2 months later, at passage 5 to 6, the cells began to grow rapidly and thereafter could be serially subcultured at a dilution of 1:2 or 1:4 every week. This new cell line was designated JN-DSRCT-1 and has been maintained in vitro for more than 190 passages (a period of more than 40 months). The population-doubling time of JN-DSRCT-1 cells in logarithmic growth phase was approximately 38 hours. The maximum cell density was approximately 5.2 × 105 cells/cm2.
Tumorigenicity in Severe Combined Immunodeficiency (SCID) Mice
To evaluate the tumorigenicity of the cell line, we injected JN-DSRCT-1 cells subcutaneously into SCID mice. Small elastic hard nodules were first palpable in SCID mice approximately 4 weeks after inoculation of JN-DSRCT-1 cells. Two months later, the tumors had grown to 1.5 cm in diameter. The cut surfaces of these tumors were solid and white with small necrotic foci.
Morphologic Characterization In Vitro and In Vivo
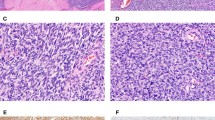
As assessed by light microscopy, JN-DSRCT-1 cells growing on coverslips were small round or spindle shaped with extended thin processes. The cells proliferated loosely or in a sheet-like pattern accompanied by irregularly piled-up foci. The nuclei were oval with inconspicuous nucleoli (Fig. 2A). As shown by immunocytochemistry (Table 2), these cells were mostly positive with antibodies against vimentin (Fig. 2B), desmin (Fig. 2C), AE1/AE3 (Fig. 2D), CAM 5.2, epithelial membrane antigen (EMA) (Fig. 2E), CD57 (Fig. 2F), and WT1 and occasionally with antibodies against neuron-specific enolase (NSE) and CD99. The other antibodies tested in vitro were negative. On the other hand, the heterotransplanted tumors revealed essentially the same histology as the original tumor, although the tumor cells had more abundant vacuolated cytoplasm or a signet ring-like appearance. Specifically, the tumors were composed of irregular clusters or strands of small round cells embedded in a dense desmoplastic stroma (Fig. 3A). Immunohistochemically (Table 2) the tumor cells were frequently positive for vimentin (Fig. 3B) and desmin (Fig. 3C) and occasionally for AE1/AE3, CAM 5.2, EMA (Fig. 3D), NSE (Fig. 3E), CD57, CD99, and WT1 (Fig. 3F) but were negative for the other antibodies tested in vivo.

Light microscopic findings of JN-DSRCT-1 cells in vitro. A, Small round or spindle shaped cells grown in a monolayer, containing oval nuclei with inconspicuous nucleoli and thin cytoplasmic processes (HE, original magnification, × 160). Most JN-DSRCT-1 cells exhibit strongly immunopositive reaction for vimentin (B), desmin (C), AE1/AE3 (D), epithelial membrane antigen (EMA; E), and CD57 (F) (streptavidin-biotin procedure, original magnification, × 160).

Light microscopic findings of JN-DSRCT-1 cells in vivo. A, A representative portion of the tumor in a SCID mouse, essentially resembling the original DSRCT (HE, original magnification, × 80). Strong and diffuse immunopositive reaction of tumor cells for vimentin (B) and desmin (C). Focally positive immunoreactivity of tumor cells for EMA (D) and neuron-specific enolase (NSE; E). F, Nuclei of tumor cells, weakly immunostained for Wilms’ tumor gene (WT1) (streptavidin-biotin procedure, original magnification, × 160).
Cytogenetic Findings
To assess the karyotype of the cell line, a total of 15 JN-DSRCT-1 metaphases were examined. All metaphases revealed the same karyotypic pattern of 48, XY, +5, der(8)del(8)(p21)t(1;8)(q21;q24), t(11;22)(p13;q12), del(16)(q22), del(17)(p11), add(21)(p11), +der(22)t(11;22) (p13;q12) (Fig. 4).

A G-banded karyotype of a metaphase JN-DSRCT-1 cell including a pathognomonic translocation, t(11;22)(p13;q12). Arrows indicate the abnormalities noted.
Molecular Findings
To verify the t(11;22)(p13;q12) translocation in the JN-DSRCT-1 cell line, we used RT-PCR to detect the EWS-WT1 fusion transcript. As shown in Figure 5, the EWS-WT1 fusion transcripts were detected as a single band of 449 bp in the JN-DSRCT-1 cells and in the original tumor tissue but not in the negative controls. However, the size of PCR products in this study was much larger than that seen characteristically in DSRCT (the expected size was 197 bp). The glyceraldehyde-3-phosphate dehydrogenase (G3PDH) transcripts from each sample yielded a single band of approximately 450 bp (Fig. 5). Nucleotide sequence analysis of the PCR products showed an in-frame fusion of EWS exon 10 to WT1 exon 8, corresponding to a variant EWS-WT1 gene fusion previously described by Antonescu et al (1998) (Fig. 6).

Detection of Ewing’s sarcoma gene (EWS)-WT1 fusion transcripts by RT-PCR. JN-DSRCT-1 cells (Lane 1) and original DSRCT tissue (Lane 2) generated products of 449 bp, corresponding to the EWS-WT1 fusion transcripts. Glyceraldehyde-3-phosphate dehydrogenase (G3PDH) transcripts from each sample exhibit a single band of approximately 450 bp. M = 100-bp DNA ladder; Lane 3 = negative control (no RNA).

Sequence of the EWS-WT1 fusion transcript junction present in JN-DSRCT-1 cells. A, Nucleotide sequence of the cDNA obtained by RT-PCR amplification of the chimeric transcript. Vertical bar indicates the position of the junction between EWS and WT1. B, Junction sequence of a variant EWS-WT1 fusion transcript. Only the portion of the exons adjacent to the junction is shown.
Discussion
To our knowledge, no human cell line derived from DSRCT has been described. In this study, we established a new human cell line, JN-DSRCT-1, from the pleural effusion of a 7-year-old boy with pulmonary metastasis. JN-DSRCT-1 cells, both in vitro and in vivo, exhibited a small round shape with immunopositive reactions for epithelial, mesenchymal, and neural markers. In addition, the JN-DSRCT-1 cells in vivo exhibited morphologic characteristics of DSRCT, such as irregular nests or clusters of small round cells embedded in a dense desmoplastic stroma. Therefore, these findings obtained from the JN-DSRCT-1 cell line were compatible with those of human DSRCT (Ordonez, 1998a, 1998b; Weiss and Goldblum, 2001).
Recurring chromosome rearrangements, particularly those that serve as diagnostic or prognostic markers, are often critical events in tumorigenesis in a number of soft tissue tumors (Weiss and Goldblum, 2001). The nature of each chromosome rearrangement often suggests the mechanism by which specific genes participate in tumorigenesis. The unique translocation of chromosomes 11 and 22, t(11;22)(p13;q12), has been detected in more than 75% of DSRCTs (Weiss and Goldblum, 2001). This translocation is therefore considered pathognomonic for this tumor and leads to the formation of a fusion gene between WT1 on chromosome 11 and EWS on chromosome 22 (Ladanyi and Gerald, 1994). Using RT-PCR, it is possible to detect the EWS-WT1 fusion transcripts (Argatoff et al, 1996; Brodie et al, 1995; de Alava et al, 1995; Gerald et al, 1995). Thus, detection of this specific translocation by cytogenetic analysis and of EWS-WT1 fusion transcripts by RT-PCR are particularly useful for identifying DSRCT, which may be difficult to distinguish from other small round cell tumors including extraskeletal Ewing’s sarcoma/primitive neuroectodermal tumor, rhabdomyosarcoma, neuroblastoma, lymphoma, poorly differentiated carcinoma, small-cell carcinoma, Merkel-cell carcinoma, and malignant mesothelioma. In this study, JN-DSRCT-1 cells showed the pathognomonic t(11;22)(p13;q12) translocation in all cells examined. Furthermore, JN-DSRCT-1 cells were proven specific for the EWS-WT1 fusion gene by RT-PCR. To our knowledge, the EWS-WT1 fusion gene has been described only in DSRCTs, although some variant forms of EWS-WT1 fusion as in our case have been reported (Adsay et al, 1999, Antonescu et al, 1998; Chan et al, 1999; Shimizu et al, 1998). Therefore, these cytogenetic and molecular genetic findings also support the JN-DSRCT-1 cell line as a DSRCT.
In conclusion, we have isolated and characterized a novel permanent human cell line JN-DSRCT-1, established from an intra-abdominal DSRCT. This cell line has retained the morphologic characteristics of DSRCT both in vitro and in vivo, with expression of epithelial, mesenchymal, and neural antigens and the EWS-WT1 fusion gene. Therefore, JN-DSRCT-1 should provide us with a new experimental system in which to study the pathogenic mechanisms and biologic behavior of human DSRCT, and importantly, to provide a cell model in which to test new therapeutic strategies and reagents against human DSRCT.
Materials and Methods
Source of Tumor Cells
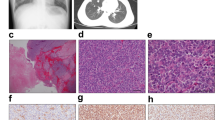
A 7-year-old Japanese boy presented with abdominal enlargement and dysuria. A large mass was palpated in the upper part of this abdomen. An open biopsy of the liver was performed, and the tumor was histopathologically diagnosed as a DSRCT (Fig. 1A). Cytogenetic analysis of short-term cultured tumor cells revealed a specific translocation, t(11;22)(p13;q12). As shown in Table 2, most of the tumor cells were strongly immunopositive for vimentin and desmin (Fig. 1B), and some were also immunopositive for EMA, cytokeratins (Fig. 1C), NSE, CD57 (Fig. 1D), and CD99. Despite intensive chemotherapy, the patient died of extensive pulmonary metastasis 26 months after the initial diagnosis. Two months before death, approximately 100 ml of the pleural effusion fluid containing tumor cells was obtained for the primary culture. No autopsy was performed.

Light microscopic findings of the original tumor primarily arising in the abdominal cavity in a 7-year-old boy. A, The original tumor is composed of irregular nests with small round cells embedded in an abundant desmoplastic stroma, corresponding to desmoplastic small round cell tumor (DSRCT) (hematoxylin and eosin [HE], original magnification, × 80). B, Strongly and diffusely immunopositive reaction of tumor cells for desmin. C, Focally positive immunoreactivity of tumor cells for AE1/AE3. D, Weakly positive immunoreactivity of some tumor cells for CD57 (streptavidin-biotin procedure, original magnification, × 160).
Establishment and Characterization of the Tumor Cell Line
After centrifugation at 1200 rpm for 10 minutes, the tumor cells were gathered from the pleural effusion. Approximately 1.0 × 107 cells were seeded in a 25-cm2 plastic flask (Falcon 3013; Becton Dickinson Japan, Tokyo, Japan) containing culture medium and maintained in a humidified atmosphere of 5% CO2 in air at 37° C. The culture medium was composed of a 1:1 mixture of DMEM and Ham’s F-12 (Kyokuto Pharmacology Company, Tokyo, Japan) supplemented with 10% FCS (Cell Culture Laboratories, Cleveland, Ohio) and kanamycin sulfate (100 μg/ml; Meiji Seika, Tokyo, Japan). The medium was replaced twice weekly. When semiconfluent layers were obtained, the cells were dispersed with PBS containing 0.1% trypsin and 0.02% EDTA solution and seeded in new flasks for passage. These procedures were serially performed until establishment of the JN-DSRCT-1 cell line.
To determine the doubling time, 1.0 × 104 JN-DSRCT-1 cells/cm2 at passage 30 were seeded in each well of 24-well dishes (Corning Costar, Tokyo, Japan) with fresh medium containing 1 ml of DMEM/F-12 with 10% FCS. The culture dishes were harvested, and then the number of viable cells in each dish was counted by the dye exclusion (0.1% trypan blue in PBS) every 24 hours for 7 days.
Tumorigenicity in SCID Mice
To determine the tumorigenicity of the JN-DSRCT-1 cell line in vivo, 5 × 107 cells at passage 24 were washed, suspended in PBS, and injected subcutaneously into the back of two 5-week-old female athymic SCID mice (CB-17/Icr-scid; Jcl Clea Japan, Inc., Osaka, Japan). The mice were maintained in a pathogen-free environment and carefully observed after transplantation. The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Fukuoka University School of Medicine.
Pathologic Studies
The cells grown in culture flasks were observed by phase-contrast microscopy. JN-DSRCT-1 cells at passage 38 were examined. For routine light microscopy, the cells cultured in chamber slides (Lab-Tek, Miles Laboratories, Naperville, Illinois) were fixed in methanol and stained with hematoxylin and eosin and periodic acid-Schiff. Paraffin sections from the original tumor and xenografts were stained with the same reagents. The primary antibodies and their dilutions used for immunocytochemistry are listed in Table 1. The cells grown in chamber slides were washed in PBS and fixed in cold acetone for 5 minutes. The cells were reacted with each of the primary antibodies for 1 hour at room temperature. The bound antibodies were then visualized using a labeled streptavidin biotin system and the alkaline phosphatase technique, as described previously (Nishio et al, 2001a). Paraffin sections from the original tumor and xenografts were also examined using the same procedure.
Cytogenetic Analysis
The JN-DSRCT-1 cells at passage 21 were used for cytogenetic analysis. Chromosome slides were prepared using the conventional trypsin-Giemsa banding technique (Nishio et al, 2001b). Karyotypes were described on the basis of the short system of the International System for Human Cytogenetic Nomenclature.
Detection of mRNA of EWS-WT1 by RT-PCR
For detection of mRNA of the EWS-WT1 fusion gene, total RNA was extracted from JN-DSRCT-1 cells at passage 21 and the original tumor tissue using Trizol reagent (GIBCO BRL, Gaithersburg, Maryland), according to previously described methods (Takeuchi et al, 1997). Reverse transcription was performed with 1 μg of total RNA in a final reaction volume of 20 μL containing Superscript II reverse transcriptase (GIBCO BRL). The reaction mixture was incubated at 42° C for 50 minutes using random hexamers. The reverse transcriptase was inactivated at 70° C for 15 minutes and on ice for 5 minutes. The PCR reagents, including 2.5 U of KOD Dash (TOYOBO Co., Osaka, Japan), were added to form a final volume of 25 μL. To verify the integrity of mRNA isolated from each sample, a parallel PCR of positive control for the ubiquitously expressed G3PDH gene was performed with the following primers: 5′-TCCACCACCCTGTTGCTGTA-3′ for sense and 5′-ACCACAGTCCATGCCATCAC-3′ for antisense. These primers amplified an approximately 450-bp fragment of G3PDH mRNA. The forward primer for EWS exon 7 was 5′-TCCTACAGCC-AAGCTCCAAGTC-3′, primer 22.3 (Delattre et al, 1992). The reverse primer for WT1 exon 9 was 5′-GACCAGGAGAACTTTCGCTGAC-3′ (Ladanyi and Gerald, 1994). PCR was then performed with an initial denaturation step of 95° C for 2 minutes, followed by 35 cycles consisting of denaturation at 94° C for 20 seconds, annealing at 57° C for 30 seconds, and extension at 72° C for 30 seconds. PCR products were resolved by electrophoresis in 3.0% agarose gels and visualized by ethidium bromide staining and ultraviolet illumination. The RT-PCR procedure was performed at least twice for each sample. Negative controls included reactions lacking RNA and reactions lacking reverse transcriptase.
Identification of Chimeric cDNA Sequences
To confirm the type of fusion gene in JN-DSRCT-1 cells, the PCR products were cloned into a pCR 4-TOPO vector using a TOPO TA cloning kit (Invitrogen Corporation, Carlsbad, California) and sequenced using a Perkin Elmer ABI Prism 377 automated sequence analyzer (Applied Biosystems, Foster City, California). Obtained sequences were compared with published data.
Availability of the Tumor Cell Line
This JN-DSRCT-1 is available from the corresponding author. Send e-mail to nishio@minf.med.fukuoka-u.ac.jp.
References
Adsay V, Cheng J, Athanasian E, Gerald WL, and Rosai J (1999). Primary desmoplastic small cell tumor of soft tissues and bone of the hand. Am J Surg Pathol 23: 1408–1413.
Antonescu DR, Gerald WL, Magid MS, and Ladanyi M (1998). Molecular variants of the EWS-WT1 gene fusion in desmoplastic small round cell tumor. Diagn Mol Pathol 7: 24–28.
Argatoff LH, O’Connell JX, Mathers JA, Gilks CB, and Sorensen PH (1996). Detection of the EWS/WT1 gene fusion by reverse transcriptase-polymerase chain reaction in the diagnosis of intra-abdominal desmoplastic small round cell tumor. Am J Surg Pathol 20: 406–412.
Biegel JA, Conard K, and Brooks JJ (1993). Translocation t(11;22)(p13;q12): Primary change in intra-abdominal desmoplastic small round cell tumor. Genes Chromosomes Cancer 7: 119–121.
Brodie SG, Stocker SJ, Wardlaw JC, Duncan MH, McConnell TS, Feddersen RM, and Williams TM (1995). EWS and WT1 gene fusion in desmoplastic small round cell tumor of the abdomen. Hum Pathol 26: 1370–1374.
Chan AS, MacNeill S, Thorner P, Squire J, and Zielenska M (1999). Variant EWS-WT1 chimeric product in the desmoplastic small round cell tumor. Pediatr Dev Pathol 2: 188–192.
de Alava E, Ladanyi M, Rosai J, and Gerald WL (1995). Detection of chimeric transcripts in desmoplastic small round cell tumor and related developmental tumors by reverse transcriptase polymerase chain reaction: A specific assay. Am J Pathol 147: 1584–1591.
Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, and Thomas G (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumors. Nature 359: 162–165.
Gerald WL, Miller HK, Battifora H, Miettinen M, Silva EG, and Rosai J (1991). Intra-abdominal desmoplastic small round-cell tumor: Report of 19 cases of distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol 15: 499–513.
Gerald WL, Rosai J, and Ladanyi M (1995). Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci USA 92: 1028–1032.
Ladanyi M and Gerald W (1994). Fusion of the EWS and WT1 genes in the desmoplastic small round-cell tumor. Cancer Res 54: 2837–2840.
Mihok NA and Cha I (2001). Desmoplastic small round cell tumor presenting as a neck mass: A case report. Diagn Cytopathol 25: 68–72.
Nishio J, Iwasaki H, Ishiguro M, Ohjimi Y, Isayama T, Naito M, and Kikuchi M (2001a). Identification of syt-ssx fusion transcripts in both epithelial and spindle cell components of biphasic synovial sarcoma in small tissue samples isolated by membrane-based laser microdissection. Virchows Arch 439: 152–157.
Nishio J, Iwasaki H, Ishiguro M, Ohjimi Y, Yoh S, Isayama T, Naito M, and Kikuchi M (2001b). Supernumerary ring chromosome in a Bednar tumor (pigmented dermatofibrosarcoma protuberans) is composed of interspersed sequences from chromosomes 17 and 22: A fluorescence in situ hybridization and comparative genomic hybridization analysis. Genes Chromosomes Cancer 30: 305–309.
Ordonez NG (1998a). Desmoplastic small round cell tumor. II: An ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. Am J Surg Pathol 22: 1314–1327.
Ordonez NG (1998b). Desmoplastic small round cell tumor. I: A histopathologic study of 39 cases with emphasis on unusual histological patterns. Am J Surg Pathol 22: 1303–1313.
Parkash V, Gerald WL, Parma A, Miettinen M, and Rosai J (1995). Desmoplastic small round-cell tumor of the pleura. Am J Surg Pathol 19: 659–665.
Rodriguez E, Sreekantaiah C, Gerald W, Reuter VE, Motzer RJ, and Chaganti RSK (1993). A recurring translocation, t(11;22)(p13;q11.2), characterizes intra-abdominal desmoplastic small round-cell tumors. Cancer Genet Cytogenet 69: 17–21.
Sawyer JR, Tyrka AT, and Lewis JM (1992). A novel reciprocal chromosome translocation t(11;22)(p13;q12) in an intra-abdominal desmoplastic small round-cell tumor. Am J Surg Pathol 16: 411–416.
Shimizu Y, Mitsui T, Kawakami T, Ikegami T, Kanazawa C, Katsuura M, Obata K, Yamagiwa I, and Hayasaka K (1998). Novel breakpoints of the EWS gene and the WT1 gene in a desmoplastic small round cell tumor. Cancer Genet Cytogenet 106: 156–158.
Takeuchi T, Iwasaki H, Ohjimi Y, Ohshima K, Kaneko Y, Ishiguro M, Hiratsuka Y, Sakamoto K, and Kikuchi M (1997). Renal primitive neuroectodermal tumor: A morphologic, cytogenetic, and molecular analysis with the establishment of two cultured cell lines. Diagn Mol Pathol 6: 309–317.
Tison V, Cerasoli S, Morigi F, Ladanyi M, Gerald WL, and Rosai J (1996). Intracranial desmoplastic small-cell tumor: Report of a case. Am J Surg Pathol 20: 112–117.
Weiss SW and Goldblum JR (2001). Malignant soft tissue tumors of uncertain type. In: Enzinger FM, and Weiss SW, editors. Soft tissue tumors, 4th ed. St Louis: Mosby, 1538–1545.
Wolf AN, Ladanyi M, Paull G, Blaugrund J, and Westra W (1999). The expanding clinical spectrum of desmoplastic small round-cell tumor: A report of two cases with molecular confirmation. Hum Pathol 1999 30: 430–435.
Acknowledgements
The authors thank Associate Professor F. G. Issa for revising the English used in this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nishio, J., Iwasaki, H., Ishiguro, M. et al. Establishment and Characterization of a Novel Human Desmoplastic Small Round Cell Tumor Cell Line, JN-DSRCT-1. Lab Invest 82, 1175–1182 (2002). https://doi.org/10.1097/01.LAB.0000028059.92642.03
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000028059.92642.03
