
Abstract
The high incidence of HER2 overexpression on the cell surface of breast cancer cells and the recognized prognostic and potentially predictive value of HER2 render this cell surface receptor a novel and important therapeutic target. Although immunohistochemistry (IHC; HercepTest) and fluorescence in situ hybridization (FISH; PathVysion and INFORM)—both approved by the Food and Drug Administration—have emerged as the most viable assays for evaluation of HER2 status in routine clinical practice, there is still no consensus on which is the best method for assessing HER2 status. Therefore, our specific objective was to establish a chromogenic in situ hybridization (CISH) assay for the detection of HER2 amplification on a cohort of 173 archival invasive breast carcinomas. Results were compared with HercepTest, which is the most frequently used method for detecting HER2 alteration. Additionally, HER2 gene copy number was investigated using differential PCR (dPCR) as a testing system. HER2 overexpression was found by IHC in 24.3%; HER2 amplification was found by CISH in 19.1% and by dPCR in 9.2% of the tumors. The overall concordance rate was 95.9% between CISH and IHC and 85.0% between dPCR and IHC. Kappa statistics revealed an excellent agreement between IHC and CISH (κ = 0.878), but only a moderate agreement was found between IHC and dPCR (κ = 0.482). Discrepant cases between CISH and HercepTest and all IHC-positive cases (+2 and +3), a total of 42 cases, were analyzed with the FISH PathVysion (Vysis) assay. Among 25 HercepTest-positive cases (score +3), 2 showed no gene amplification by FISH or CISH. Four of 13 tumors with weak HER2 overexpression (score +2) were negative with both FISH and CISH. Concordance between CISH and FISH was 100% for the 38 cases analyzed. The current study showed that CISH represents a practical and simple assay for evaluating HER2 gene amplification in archival material, offering a promising alternative to IHC or FISH for the routine diagnostic setting.
Similar content being viewed by others

Introduction
Amplification of the HER2 gene is an important alteration in human breast cancer occurring in 25% to 30% of invasive ductal breast carcinomas (Slamon et al, 1989) and has been associated with a significant reduction in disease-free and overall survival, especially in node-positive patients (Press et al, 1997; Seshadri et al, 1993; Slamon et al, 1987; Toikkanen et al, 1992; Zhou et al, 1989). More recently, there has been considerable interest in the potential role of HER2 alteration as a predictor of response or resistance to various therapeutic modalities in patients with breast cancer. In particular, HER2 amplification and overexpression have been shown to correlate with benefit from adjuvant doxorubicin and lack of benefit from adjuvant tamoxifen and cyclophosphamide, methotrexate, and fluorouracil (Baselga et al, 1998; Cobleigh et al, 1999; Paik et al, 1998). With the availability of the humanized monoclonal antibody trastuzumab (Herceptin; Genentech, Inc., South San Francisco, California), which is directed against the extracellular part of the HER2 receptor protein, there has been growing clinical demand for HER2 analysis of current and archived breast cancer specimens. Optimal use of this therapy requires accurate determination of HER2 status because, to date, the presence of this alteration is the sole criterion for Herceptin eligibility. Fluorescence in situ hybridization (FISH; INFORM from Oncor/Ventana [Tucson, Arizona] and PathVysion from Vysis [Downers Grove, Illinois]) and immunohistochemistry (IHC, DAKO HercepTest; DAKO, Glostrup, Denmark) methods have been approved by the U.S. Food and Drug Administration (FDA) and have emerged as the most viable assays for evaluation of HER2 status in routine clinical practice. Each of these methods has advantages but also disadvantages. Thus, up to now, consensus regarding the best methods, reagents, or cut-off points to define HER2 status for determining Herceptin responsiveness has not yet been reached (Thor, 2001).
From the practical standpoint, IHC staining is the most frequently used method for the assessment of HER2 status, being available as a standard technique in all pathology laboratories (Birner et al, 2001; Thor, 2001). However, assay reliability has been questioned, because specimen handling, fixation, cell conditioning, and objective scoring can affect the quality of results (Jacobs et al, 1999; Press et al, 1994; van de Vijver, 2001). Furthermore, major concerns have been raised with respect to possible oversensitivity of the DAKO HercepTest, resulting in false-positive results (Jacobs et al, 1999; Tubbs et al, 2001). Although the FISH procedure has been proven to be reliable and reproducible (Bartlett et al, 2001; Pauletti et al, 1996; Tubbs et al, 2001), a colorimetric or chromogenic modification of this assay would be highly desirable for most pathologists, who are more familiar and experienced with a brightfield microscope, which can be used for evaluation of colorimetric assays. More recently, a chromogenic in situ hybridization (CISH) has been introduced by Tanner and coworkers (Tanner et al, 2000) as an alternative to FISH, in which the DNA probe is detected using a peroxidase reaction. Others (Sharma et al, 1999; Smith et al, 1994; Vos et al, 1999) have also successfully used nonfluorescent ISH for the detection of HER2 oncogene.
Therefore, our specific objective was to establish a CISH assay for the detection of HER2 amplification in routinely processed archival human breast carcinomas. This technique was compared with the DAKO HercepTest, which is the standard technique for detecting HER2 protein overexpression not only in our department but also in many other pathology departments (Thor, 2001). We also investigated the HER2 gene copy number in the tumors using differential PCR (dPCR) as a test system. Finally, discrepant cases between CISH and HercepTest and all IHC positive cases (+2 and +3), a total of 42 cases, were analyzed with the FISH PathVysion (Vysis) assay.
Results
IHC (HercepTest)
The results of immunostaining of invasive breast cancers for HER2 overexpression by means of the HercepTest are listed in Table 1. The expression of HER2 was found to be negative in 131 (75.7%) of 173 cases, weak in 17 (9.8%) of 173 cases, and strong in 25 (14.5%) of 173 cases. Using the HercepTest scoring system, 42 (24.3%) cases were interpreted as positive (score +2 or +3), and 131 cases (75.7%) were interpreted as negative (score of 0 or +1).
CISH
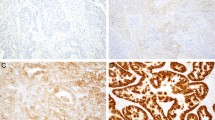
One hundred seventy-one of the 173 cases under investigation were assessable for CISH. Two cases had insufficient tumor present on the slides. Positive hybridization signals were clearly distinguished using × 25 to × 40 objectives in the nuclei of tumor cells. Amplified gene copies appeared typically as numerous individual brown chromogenic reaction product signals (Fig. 1A) or as brown intranuclear gene copy clusters (Fig. 1B). Tumors with no amplification showed typically one to two spots per nucleus (Fig. 1C) (when diploid) or two to four spots in cases of chromosomal aneuploidy (Fig. 1D). Of 171 investigated cases, 138 (79.8%) cases showed no amplification and 33 cases (19.1%) revealed a HER2 amplification (Table 1).

Representative photomicrographs of HER2 oncogene detection determined by chromogenic in situ hybridization (CISH) in archival invasive breast carcinomas. Tumor with high level of HER2 gene amplification showing either individual scattered brown reaction products (A) or numerous signals, arranged in clusters (B). Tumor with no amplification shows typically one to two signals per nucleus (C) or two to four spots in a case of chromosomal aneuploidy (D). An invasive ductal carcinoma with HER2 low-level amplification by CISH (E) and by fluorescence in situ hybridization (FISH) (F). Counterstained with hematoxylin (A to E) and 4,6-diamidino-2-phenylindole (F); magnification × 405 (A to E) and × 1013 (F).
dPCR
To validate dPCR, DNA extracted from formalin-fixed, paraffin-embedded normal placenta and different human breast cancer cell lines was tested to determine the sensitivity, accuracy, and reproducibility of this method in quantifying HER2 gene amplification. The mean ratio values ± sd achieved for the breast cancer cell lines BT474, SKBR3, MDA MB 361, HBL 100, BT 20, ALAB, and for normal human placenta were 1.39 ± 0.34, 1.08 ± 0.20, 0.64 ± 0.15, 0.14 ± 0.07, 0.20 ± 0.06, 0.13 ± 0.03, and 0.13 ± 0.05, respectively. The ratios of HER2 to IFN-γ showed a linear relation with the number of copies of HER2 present when multiple samples from each cell line were analyzed (data not shown).
The ratio values obtained for all 173 breast cancer tissues fell between 0.03 and 5.79 (mean 0.31, sd 0.67). This mean value corresponds to approximately 2.4 gene copies. A total of 157 tumors had ratio values ranging between 0.01 and 0.56 (mean 0.16, sd 0.12) and were categorized as negative (≤4 copies). Sixteen of the 173 tumors analyzed demonstrated a HER2 gene amplification (9.2%) and had ratio values between 0.73 and 5.79 (mean 1.75, sd 1.6) (Table 1). This mean value corresponds to approximately 13.5 gene copies. Figure 2 shows the results of PCR analyses of four representative cases.

Analysis of HER2 gene amplification by differential PCR (dPCR) from four cases of archival invasive breast carcinomas (lanes 5 to 8). MW = molecular weight marker; lane 1 = negative control; lane 2 = BT474; lane 3 = SKBR3; lane 4 = normal placenta. Lanes 2, 3, and 5 show clear amplification of the HER2 product.
Correlation of IHC, CISH, and dPCR Measurement for HER2 Status
The results of both IHC and CISH assays were assessable in 171 cases (Table 2). Of the 171 studied cases, 131 (76.6%) tumors showed no gene amplification and no HER2 protein overexpression; 22 (12.9%) tumors showed strong HER2 protein overexpression and HER2 amplification by CISH; and 11 (6.4%) tumors demonstrated weak HER2 protein overexpression and gene amplification by CISH. Five (2.9%) tumors with weak HER2 protein overexpression were found to be negative by CISH. Two (1.2%) tumors revealed strong HER2 overexpression but no gene amplification by CISH. Overall concordance between CISH and IHC was found in 164 (95.9%) of 171 cases. All tumors that showed no HER2 gene amplification by dPCR (131/173) (Table 2) were also negative for HER2 protein overexpression by IHC. Fourteen (8.1%) IHC weakly positive and 12 (6.9%) IHC strongly positive cases were negative with dPCR. Overall concordance between dPCR and IHC was obtained in 147 (85.0%) of 173 cases. κ statistics revealed an excellent agreement between IHC and CISH (κ = 0.878). In contrast, only a moderate agreement was found between dPCR and IHC (κ = 0.482) (Table 3).
FISH
Discrepant cases between CISH and HercepTest (7/171) and all IHC-positive cases (+2 and +3), a total of 42 cases, were analyzed with the FISH (PathVysion) assay (Table 4). Of these 42 cases, 2 cases could not be analyzed with FISH because they had insufficient tumor present on the slides and a further 2 cases could not be scored because of high background and low signal intensity, despite several trials. Among 25 HercepTest-positive tumors (score of 3), 2 cases showed no gene amplification by FISH or by CISH. Four of 13 tumors with weak HER2 overexpression (score +2) were categorized as nonamplified with FISH or CISH. Concordance between CISH and FISH was 100% for the 38 cases analyzed (Fig. 1, E and F).
Discussion
Beyond its prognostic and potentially predictive value, HER2 is a highly promising and specific anticancer target, and therefore there is a growing clinical demand for HER2 analysis on current and archival breast cancer specimens, especially with the introduction of Herceptin. Optimal use of this therapy requires accurate determination of HER2 status because, to date, the presence of this alteration is the sole criterion for Herceptin eligibility (Shak, 1999). Although FISH (INFORM from Oncor/Ventana and PathVysion from Vysis) and IHC (DAKO HercepTest) methods have been approved by the U.S. FDA as clinical tests for breast carcinomas, there is still no consensus as to which is the most reliable and accurate method to assess HER2 status, especially in the context of Herceptin therapy (Thor, 2001). Although FISH is currently considered to be the most specific and sensitive method for detection of oncogene amplification in human tumor samples, most pathologists may favor a colorimetric modification of this assay, which allows histopathologic evaluation of tissues using a standard brightfield microscope.
The present study was therefore designed to establish a CISH technique for the detection of HER2 alteration in routinely processed archival human breast carcinomas. Compared with FISH, CISH offers the possibility of simple and fast signal evaluation of tumor cells on a cell-to-cell basis using a standard brightfield microscope, simultaneous morphologic examination of histopathology, and permanent storage of slides. Using a cohort of 173 archival breast cancer specimens, we demonstrated that CISH is an assay that is simple to use and can be easily integrated into routine testing. Gene amplification by CISH was easy to identify under the microscope with a × 25 or × 40 objective. Two different signal distribution patterns have been observed, either consistent signal clusters or individual scattered signals. Signal distribution as groups or clusters has been described previously by others using FISH for the detection of HER2 (Couturier et al, 2000; Pauletti et al, 1996; Press et al, 1997). Even though double minute chromosomes have been shown to have a tendency to aggregate, this pattern is more likely a result of intrachromosomal amplification, namely homogeneously staining regions (Cowell, 1982). Precise signal enumeration of high-level amplification was frequently not possible because of coalescing signal clusters, but at least in routine diagnostics, enumeration of gene copies exceeding 10 is not needed. The most difficult category in CISH is the low-level amplification (six to eight copies), in which accurate enumeration of the gene copies is necessary.
The current CISH procedure is based on a single-color detection of one probe, similar to the FDA-approved FISH test (INFORM; Oncor/Ventana). The theoretical advantage of two-colored FISH (PathVysion; Vysis) is its ability to distinguish chromosomal amplification from aneuploidy using a chromosome 17 centromere as a differentially labeled reference probe. The use of chromosome 17 correction has been suggested as an important means to correct for HER2 pseudoamplification caused by chromosome 17 polysomy (Paik et al, 1998; Press et al, 1997). Conversely, others (Jimenez et al, 2000; McManus et al, 1999; Tubbs et al, 2001) reported that although chromosome 17 centromere counts do not provide additional information, they make analysis even more time consuming.
Tanner and coworkers (Tanner et al, 2000) recently introduced a CISH assay as a practical alternative to FISH. They used a digoxigenin-labeled probe from Zymed, which was subsequently detected with a two-step technique using anti-digoxigenin fluorescein and anti-fluorescein peroxidase. CISH results were then compared with FISH. However, it is important to note that these authors have used different sample materials, which might explain the lower sensitivity and some of the discrepancies between these methods. Although FISH was performed on entire nuclei derived from fresh-frozen tissue sections, CISH was performed on archival formalin-fixed and paraffin-embedded tissues. In contrast, we have used identical sample material—archival formalin-fixed and paraffin-embedded tissue specimens—for IHC, CISH, and dPCR. Furthermore, detection of the probe was performed using a three-step signal amplification technique consisting of mouse anti-digoxigenin, biotinylated goat anti-mouse, and a horseradish-streptavidin-biotin complex, allowing for a method with higher sensitivity.
In the current study, we evaluated IHC, CISH, and dPCR concurrently in a cohort of 173 invasive breast carcinomas. A total of 42 (24.3%) of 173 investigated tumors displayed positive (+2, +3) immunostaining and 131 (75.7%) cases were scored as negative (0, +1) with IHC. In comparison, 33 (19.1%) of 171 patients in this same cohort demonstrated HER2 gene amplification and 138 (79.8%) cases were negative with CISH. HER2 gene alteration determined by dPCR, however, was detectable in 16 (9.2%) of 173 cases, and 157 (90.8%) tumors showed no HER2 gene amplification with dPCR. The HER2 overexpression/amplification rates determined by IHC (24.3%) and CISH (19.1%) are in concordance with established measures in breast cancers (Press et al, 1997; Slamon et al, 1987, 1989). The 9.2% amplification frequency, detected by dPCR, somewhat underestimates this prevalence. However, it is well recognized that PCR-based assays suffer from lower sensitivity and dilution artifacts because of variations in tumor cell content within the given specimen (Slamon et al, 1989). Optimal length of probes used for ISH techniques is within the range of 50 to 300 bp (Hoffler et al, 1998). Although longer probes allow for a higher sensitivity, nonspecific hybridization occurs more frequently with longer probes (>100 bp). In contrast, with PCR, very short primers (20–30 bp) are used that on the one hand are highly specific but on the other hand lack sensitivity. Thus, this fact might be another explanation for the relatively low sensitivity of dPCR.
In our series, the overall concordance was 95.9% between CISH and IHC and 85.0% between dPCR and IHC. Furthermore, κ statistics revealed an excellent agreement between IHC and CISH (κ = 0.878). In contrast, only a moderate agreement was found between dPCR and IHC (k = 0.482). These findings, however, confirm that sensitivity of PCR-based assays suffers from the problem of variation in tumor cell content within a given specimen. Significant misinterpretation of data can result from these techniques because of the dilution of malignant cells by normal stroma, vascular, or inflammatory cells contained within the tumor specimen. This is particularly significant in breast cancer, because nonmalignant cells might constitute more than 50% of the cells in a sample (Slamon et al, 1987). Thus, this fact is very likely to account for the low percentage of positive cases (9.2%) determined by dPCR. Therefore, in our opinion the use of dPCR in determining HER2 amplification as a routine method for clinical application is neither suitable nor recommended. However, improvement in the molecular analysis for gene amplification evaluation can be attained by using microdissected tumor material and real-time PCR instead of competitive or differential methods, because it allows better internal control, less manipulation of the specimen, and objective quantitative results (Bieche and Lidereau 1995; Heid et al, 1996). Yet, this technology is not widely available to all laboratories.
Finally, discrepant cases between CISH and HercepTest (7/171) and all IHC-positive cases (+2 and +3), a total of 42 cases, were analyzed with the FISH (PathVysion) assay. With respect to those tumors defined as nonamplified by CISH, 7 (4.1%) cases of 171 tumors showed HER2 overexpression in the absence of gene amplification. These 7 IHC-positive cases were also classified as nonamplified using FISH. Concordance between CISH and FISH was 100% for the 38 cases analyzed. Thus, this CISH technique seems to be sensitive and specific for detection of HER2 amplification in human archival tumor samples. However, HER2 is a growth factor, and enhanced transcription in the absence of gene amplification is a well-recognized mechanism for cellular function through enhanced production of mRNA by phosphorylation of tyrosine kinase acting on growth factors and regulators of cell growth and proliferation (Earp et al, 1995; Pauletti et al, 1996). Thus, discordances between HER2 overexpression and amplification may simply reflect this fact.
CONCLUSION
In conclusion, the current study showed that CISH represents an interesting assay for evaluating HER2 gene alteration in routinely processed and archival material. CISH is able to overcome several technical and practical limitations inherent to FISH and IHC and thus might provide a promising new approach to identifying HER2 alteration in routine clinical use.
Materials and Methods
Tumor Specimens
One hundred seventy-three cases of invasive breast carcinomas diagnosed between 1993 and 1999 were obtained from the Institute of Pathology, Landeskliniken Salzburg, Austria. Tumor tissue was routinely processed for (immuno)histologic analysis. Formalin-fixed, paraffin-embedded tumor blocks were available for all patients. Histologic classification was performed according to the World Health Organization guidelines, and tumor staging was performed according to the TNM System of the International Union against Cancer. The total of 173 cases examined comprised 123 (71.1%) infiltrating ductal carcinomas, 27 (15.6%) infiltrating lobular carcinomas, 8 (4.6%) mixed infiltrating ductal and lobular carcinomas, and 15 (8.7%) other types. Histologic grading was assessed according to Bloom and Richardson criteria. Twelve tumors (6.9%) were classified as grade 1, 116 (67.1%) as grade 2, and 45 (26.0%) as grade 3. Eighty-seven women (50.3%) were lymph node negative and 86 (49.7%) were lymph node positive.
Human Breast Cancer Cell Lines
In this study, the following established mammary carcinoma cell lines were used: HBL100, MDA MB 361, BT 20, ALAB, and SKBR3 (gift from the University Hospital of Innsbruck, Institute of Pathology, Innsbruck, Austria). The human nontumorigenic myoepithelial HBL 100 cell line and mammary carcinoma cells were cultured in RPMI (Gibco BRL, Paisley, United Kingdom) supplemented with 10% heat-inactivated FCS (Gibco BRL), 2 mm l-glutamine, and 2 mm penicillin/streptomycin (Sigma-Aldrich, Vienna, Austria). BT474 cells were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and were cultured according to the protocol supplied.
Protein-Based Assay (HercepTest)
HER2 overexpression was assessed using the DAKO HercepTest kit (DAKO, Glostrup, Denmark). Staining and scoring were performed according to the protocol described in the manufacturer’s guidelines.
CISH
CISH was performed on 4-μm thick archival formalin-fixed paraffin-embedded tumor samples. Sections were deparaffinized and then transferred to distilled water. The tissue sections then underwent cell conditioning in a 95° C water bath; they were immersed in epitope retrieval solution (DAKO) for 20 minutes and then allowed to cool at room temperature. Enzymatic digestion was performed by incubating sections with Proteinase K (1:2000 dilution; DAKO) for 8 minutes at room temperature. The enzymatic action was stopped in distilled water, endogenous peroxidase activity was blocked with 3% H2O2 in methanol, and sections were then allowed to air dry. Fifteen microliters of the ready-to-use digoxigenin-labeled HER2 probe (Zymed, South San Francisco, California) were applied to the sections, which were then covered under coverslips. The slides were denatured on a hot plate (94° C) for 5 minutes, and hybridization was performed overnight at 37° C. To remove the coverslips, the slides were soaked in 1× Tris-buffered saline containing 0.1% Tween 20. Slides were then washed with a stringent wash solution (DAKO GenPoint, Glostrup, Denmark) at 55° C for 20 minutes. Sections were blocked with normal serum and subsequently followed by incubation with mouse anti-digoxigenin for 60 minutes at room temperature (1:200 dilution; Boehringer Mannheim, Mannheim, Germany), biotinylated goat anti-mouse (30 minutes at room temperature; 1:200 dilution), and horseradish-streptavidin-biotin complex (30 minutes at room temperature; 1:300 dilution; DAKO StreptABComplex/HRP Duet kit; DAKO). Slides were counterstained with hematoxylin.
Control tissue sections were hybridized in the absence of probe. Positive controls were cases of breast carcinoma shown to have high levels of HER2 amplification by Southern blot analysis.
Interpretation was performed independently by two of the authors (N.D. and C.H.-K.), each blinded to one another and to the results of the other assays. Hybridization signals from at least 60 tumor cells were scored to assess oncogene copy number. The probe displays a single distinct small dark brown signal at the location of each copy of the HER2 gene. The expected number of signals in a normal and in an unamplified tumor cell varies from two to four, depending on the phase of the cell cycle, and was classified as negative. Precise signal enumeration was not possible in some sections because tumors with high levels of gene amplification often exhibit coalescing signal clusters. Thus, amplification was defined as more than six signals per nucleus or when gene copy clusters were seen in >50% of cancer cells.
PathVysion (Vysis)
For the PathVysion assay, the HER2 DNA probe kit and the paraffin wax pretreatment reagent kit were purchased from Vysis (Downers Grove, Illinois). The FISH assay was performed according to the manufacturer’s recommended protocol.
In each specimen, at least 60 cells were counted for both HER2 gene and chromosome 17 centromere signals under oil immersion at × 1000 magnification using the recommended filters. Specimens with a signal ratio of less than 2.0 were designated as nonamplified and 2.0 or greater as amplified.
DNA Extraction
DNA was isolated from cells and from paraffin-embedded tissue using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Briefly, paraffin blocks were cut in two to three sections at 10 to 30 μm (depending on tissue size), deparaffinized in xylene and absolute ethanol, treated with Proteinase K, and purified on QIAamp spin columns. The eluted DNA was tested for concentration and quality by measuring absorbance at 260 nm and determining the A260/A280 ratio using a GeneQuant spectrophotometer (Pharmacia Biotech, St. Albans, Herts, United Kingdom). All DNA was diluted to the same concentration (40 ng/μl).
dPCR
One hundred seventy-three formalin-fixed, paraffin-embedded tissue sections were analyzed by dPCR. dPCR is based on the protocol originally published by Frye and coworkers (Frye et al, 1989). A 98-bp fragment of the HER2 oncogene was coamplified in the same reaction tube, with a 150-bp segment of the IFN-γ gene serving as an internal standard. The following primers were used in this study: HER2 (98 bp): 5′primer CTC TGA CGT CCA TCA TCT C, 3′primer ATC TTC TGC TGC CGT CGC TT; IFN-γ (150 bp): 5′primer TCT TTT CTT TCC CGA TAG GT, 3′primer CTG GGA TGC TCT TCG ACC TC. All oligonucleotides were synthesized by MWG-Biotech AG (Ebersberg, Germany).
dPCR was performed in duplicate for each. Total reaction volume was 50 μl containing 25 μl of Qiagen PCR master mix (Qiagen, Hilden, Germany) (1.5 mm MgCl2, 200 μm of each dNTP, 2.5 U of Taq DNA polymerase), 1 μl of each primer (0.5 μm), 5 μl of DNA (200 ng), and 16 μl of sterile distilled water. PCR cycles included 5 minutes at 95° C, then 30 cycles of 1 minute at 95° C, 1 minute at 55° C, 1 minute at 72° C, and finally 7 minutes at 72° C. A negative control was provided by omission of the DNA template. Twenty microliters of the PCR product were run on a 3% composite gel (three parts Nusieve agarose gel and one part multipurpose agarose) and visualized by ethidium bromide. The gel images were captured using a CCD camera linked to an imaging processing system (GeldocII; BioRad, Hemel Hempstead, United Kingdom). HER2 gene amplification was quantified by comparing the relative intensity of the two bands of each lane determined by means of QuantityOne (BioRad) software.
The results were expressed as the ratio of the relative intensity of the reference gene band to the relative intensity of the target gene band. In each PCR experiment, DNA extracted from formalin-fixed, paraffin-embedded normal placenta as reference tissue with nonamplified HER2 gene and the breast cancer cell lines BT474 and SKBR3 with known HER2 gene amplification status were included as positive controls.
The ratio of the signal of HER2 (A) to that of IFN-γ (B) in DNA from tumor tissue was compared with the ratio of the signal of HER2 (C) to that of IFN-γ (D) from normal placenta (one HER2 copy). Sample copy number was calculated using the formula:
copy number = (A/B)/(C/D)
A greater than 4-fold increase in copy number was considered to be amplified.
Statistical Analysis
Statistical analyses were performed using the SPSS statistical package (SPSS Inc., Chicago, Illinois). Kappa statistics were used to assess agreement between pairwise grouped methods. The κ statistic evaluates level of agreement adjusted for agreement expected to occur by chance alone. Kappa statistics less than 0.4 represent fair to poor agreement, values of 0.4 to 0.8 represent moderate to good agreement, and values of more than 0.8 represent excellent agreement.
References
Bartlett JM, Going JJ, Mallon EA, Watters AD, Reeves JR, Stanton P, Richmond J, Donald B, Ferrier R, and Cooke TG (2001). Evaluating HER2 amplification and overexpression in breast cancer. J Pathol 195: 422–428.
Baselga J, Norton L, Albanell J, Kim YM, and Mendelsohn J (1998). Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 58: 2825–2831.
Bieche I and Lidereau R (1995). Genetic alterations in breast cancer. Genes Chromosomes Cancer 14: 227–251.
Birner P, Oberhuber G, Stani J, Reithofer C, Samonigg H, Hausmaninger H, Kubista E, Kwasny W, Kandioler-Eckersberger D, Gnant M, and Jakesz R (2001). Evaluation of the United States Food and Drug Administration-approved scoring and test system of HER-2 protein expression in breast cancer. Clin Cancer Res 7: 1669–1675.
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, and Slamon DJ (1999). Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol 17: 2639–2648.
Couturier J, Vincent-Salomon A, Nicolas A, Beuzeboc P, Mouret E, Zafrani B, Sastre-Garau X, Cowell JK, Earp HS, Dawson TL, Li X, and Yu H (2000). Strong correlation between results of fluorescent in situ hybridization and immunohistochemistry for the assessment of the ERBB2 (HER-2/neu) gene status in breast carcinoma. Mod Pathol 13: 1238–1243.
Cowell JK (1982). Double minutes and homogeneously staining regions: Gene amplification in mammalian cells. Annu Rev Genet 16: 21–59.
Earp HS, Dawson TL, Li X, and Yu H (1995). Heterodimerization and functional interaction between EGF receptor family members: A new signaling paradigm with implications for breast cancer research. Breast Cancer Res Treat 35: 115–132.
Frye RA, Benz CC, and Liu E (1989). Detection of amplified oncogenes by differential polymerase chain reaction. Oncogene 4: 1153–1157.
Heid CA, Stevens J, Livak KJ, and Williams PM (1996). Real time quantitative PCR. Genome Res 6: 986–994.
Hoffler H, Mueller J, and Werner M (1998). In situ hybridization: Principles and practice. In: Polak A, and McGee JOD, editors. Principles of in situ hybridization. Oxford: Oxford Medical Publications, 1–19.
Jacobs TW, Gown AM, Yaziji H, Barnes MJ, and Schnitt SJ (1999). Comparison of fluorescence in situ hybridization and immunohistochemistry for the evaluation of HER-2/neu in breast cancer. J Clin Oncol 17: 1974–1982.
Jimenez RE, Wallis T, Tabasczka P, and Visscher DW (2000). Determination of Her-2/Neu status in breast carcinoma: Comparative analysis of immunohistochemistry and fluorescent in situ hybridization. Mod Pathol 13: 37–45.
McManus DT, Patterson AH, Maxwell P, Humphreys MW, Anderson NH, Jimenez RE, Wallis T, Tabasczka P, and Visscher DW (1999). Fluorescence in situ hybridization detection of erbB2 amplification in breast cancer fine needle aspirates. Mol Pathol 52: 75–77.
Paik S, Bryant J, Park C, Fisher B, Tan-Chiu E, Hyams D, Fisher ER, Lippman ME, Wickerham DL, and Wolmark N (1998). erbB-2 and response to doxorubicin in patients with axillary lymph node-positive, hormone receptor-negative breast cancer. J Natl Cancer Inst 90: 1361–1370.
Pauletti G, Godolphin W, Press MF, and Slamon DJ (1996). Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene 13: 63–72.
Press MF, Bernstein L, Thomas PA, Meisner LF, Zhou JY, Ma Y, Hung G, Robinson RA, Harris C, El-Naggar A, Slamon DJ, Phillips RN, Ross JS, Wolman SR, and Flom KJ (1997). HER-2/neu gene amplification characterized by fluorescence in situ hybridization: Poor prognosis in node-negative breast carcinomas. J Clin Oncol 15: 2894–2904.
Press MF, Hung G, Godolphin W, and Slamon DJ (1994). Sensitivity of HER-2/neu antibodies in archival tissue samples: Potential source of error in immunohistochemical studies of oncogene expression. Cancer Res 54: 2771–2777.
Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, and Kitchen P (1993). Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J Clin Oncol 11: 1936–1942.
Shak S (1999). Overview of the trastuzumab (Herceptin) anti-HER2 monoclonal antibody clinical program in HER2-overexpressing metastatic breast cancer. Herceptin Multinational Investigator Study Group. Semin Oncol 26: 71–77.
Sharma A, Pratap M, Sawhney VM, Khan IU, Bhambhani S, and Mitra AB (1999). Frequent amplification of C-erbB2 (HER-2/Neu) oncogene in cervical carcinoma as detected by non-fluorescence in situ hybridization technique on paraffin sections. Oncology 56: 83–87.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, and McGuire WL (1987). Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182.
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, and Ullrich A (1989). Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244: 707–712.
Smith KL, Robbins PD, Dawkins HJ, Papadimitriou JM, Redmond SL, Carrello S, Harvey JM, and Sterrett GF (1994). c-erbB-2 amplification in breast cancer: Detection in formalin-fixed, paraffin-embedded tissue by in situ hybridization. Hum Pathol 25: 413–418.
Tanner M, Gancberg D, Di Leo A, Larsimont D, Rouas G, Piccart MJ, and Isola J (2000). Chromogenic in situ hybridization: A practical alternative for fluorescence in situ hybridization to detect HER-2/neu oncogene amplification in archival breast cancer samples. Am J Pathol 157: 1467–1472.
Thor A (2001). HER2: A discussion of testing approaches in the USA. Ann Oncol 12 (Suppl 1): S101–S107.
Toikkanen S, Helin H, Isola J, and Joensuu H (1992). Prognostic significance of HER-2 oncoprotein expression in breast cancer: A 30-year follow-up. J Clin Oncol 10: 1044–1048.
Tubbs RR, Pettay JD, Roche PC, Stoler MH, Jenkins RB, and Grogan TM (2001). Discrepancies in clinical laboratory testing of eligibility for trastuzumab therapy: Apparent immunohistochemical false-positives do not get the message. J Clin Oncol 19: 2714–2721.
van de Vijver MJ (2001). Assessment of the need and appropriate method for testing for the human epidermal growth factor receptor-2 (HER2). Eur J Cancer 37 (Suppl 1): 11–17.
Vos CB, Ter Haar NT, Peterse JL, Cornelisse CJ, and van de Vijver MJ (1999). Cyclin D1 gene amplification and overexpression are present in ductal carcinoma in situ of the breast. J Pathol 187: 279–284.
Zhou DJ, Ahuja H, and Cline MJ (1989). Proto-oncogene abnormalities in human breast cancer: c-ERBB-2 amplification does not correlate with recurrence of disease. Oncogene 4: 105–108.
Acknowledgements
This work was supported in part by the Austrian National Bank Fund (grant number 7645). N.D. received a scholarship from the Medizinische Forschungsgesellschaft Salzburg, Landeskliniken Salzburg Austria, and the Österreichische Krebshilfe, Salzburg, Austria.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dandachi, N., Dietze, O. & Hauser-Kronberger, C. Chromogenic In Situ Hybridization: A Novel Approach to a Practical and Sensitive Method for the Detection of HER2 Oncogene in Archival Human Breast Carcinoma. Lab Invest 82, 1007–1014 (2002). https://doi.org/10.1097/01.LAB.0000024360.48464.A4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000024360.48464.A4
This article is cited by
-
Dual color chromogenic in situ hybridization for determination of HER2 status in breast cancer: a large comparative study to current state of the art fluorescence in situ hybridization
BMC Clinical Pathology (2012)
-
Molekulare Methoden in der Sarkomdiagnostik
Der Pathologe (2011)
-
Simultaneous detection of TOP2A and HER2 gene amplification by multiplex ligation-dependent probe amplification in breast cancer
Modern Pathology (2010)
-
Combinatorial biomarker expression in breast cancer
Breast Cancer Research and Treatment (2010)
-
Quantitative real-time RT-PCR and chromogenic in situ hybridization: precise methods to detect HER-2 status in breast carcinoma
BMC Cancer (2009)
