
Abstract
Specific gene fusions observed in solid tumors are extremely useful diagnostic markers. We report the development of a method based on real-time PCR which enables the detection upon identical PCR conditions of the different fusions specifically observed in Ewing tumors (ET), alveolar rhabdomyosarcoma (ARMS), synovial sarcoma (SS), small round cell desmoplastic tumors (SRCDT), extraskeletal myxoid chondrosarcoma, malignant melanoma of soft parts, congenital fibrosarcoma, and anaplastic large cell lymphoma. A simple assay, based on multiplexing of primers and probes, is described for the routine genetic diagnosis of small round cell tumors of children. It enables the detection of the five EWS-ETS, the two PAX-FKHR, the three SYT-SSX, and the EWS-WT1 fusions of ET, ARMS, SS, and SRCDT, respectively. The sensitivity of this test is high enough to detect all fusions, including the large EWS-FLI-1 transcripts, with the equivalent of 100 tumor cells as a starting material. This multiplex fluorescent analysis of chromosome translocations (MFACT) was validated in comparison with conventional RT-PCR on a series of 79 tumors. A major advantage of this method is that it completely abolishes the manipulation of PCR-products. It, therefore, considerably lowers the risk of cross-contamination linked to carry-over of RT-PCR products. It also constitutes an important step toward the complete automation of the detection of cancer-specific gene fusions.
Similar content being viewed by others
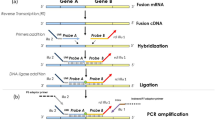


Introduction
Some solid tumors are characterized by specific translocations that result in gene fusions. These genetic lesions, which are at the basis of the tumorigenic process, now constitute very powerful diagnostic criteria (Barr, 1998; Bennicelli and Barr, 1999; Ladanyi and Bridge, 2000). Most of these gene fusions are listed in Table 1. These tumor markers are particularly useful for the precise diagnosis of sarcomas and small round cell tumors of children and young adults, which can harbor atypical clinical or pathological presentations. Different techniques, including conventional cytogenetics, Southern blotting, fluorescent in situ hybridization, reverse transcription-polymerase chain reaction (RT-PCR), or, more rarely, immunohistochemistry, have been developed to identify these lesions, with RT-PCR being the most widely used approach. Indeed, RT-PCR is a simple, specific, and sensitive technique for analyzing small tumor fragments. However, as with all PCR-based approaches, it is particularly sensitive to the risk of cross-contamination linked to the carry-over of PCR products.
We have developed real-time PCR detections of the different fusions listed in Table 1. Moreover, taking advantage of multiplexing primers and probes, we set up a test that detects the most frequent fusions observed in sarcomas and small round cell tumors of children that raise difficult diagnostic challenges.
Results
Detection of Single Gene Fusions Using Real-Time PCR
For each gene fusion listed in Table 1, internal probes and primers were designed using Primer Express software (Applied Biosystems, Foster City, California). Our aims were (a) to detect every type of gene fusion associated with a given malignancy, (b) to reach a high sensitivity of detection, and (c) to standardize PCR conditions to facilitate routine analysis.
We first focused on the detection of the various EWS-ETS fusions observed in Ewing tumor (Table 1). Concerning EWS-FLI-1, the diversity of the position of the breakpoints with respect to the exons of EWS and FLI-1 leads to an important variability of the types of fusion transcripts observed in tumors (Zucman et al, 1993b). The most proximal breakpoint observed within the EWS gene lies at codon 205 (Peter et al, 1996). Therefore, we used the primer EWS 3 tqm and the probe EWS S2 tqm, which correspond to sequences of EWS proximal to this codon. For FLI-1, fusions always contain the exon 9, which encodes the DNA binding domain. Primer FLI 3 tqm, localized within this exon, was therefore used. This set of primers and the probe would be sufficient to amplify all types of EWS-FLI-1 fusion transcripts of Ewing tumors. Initial experiments were performed using RNAs from the POE cell line, which expresses the most frequent type 1 EWS-FLI-1 fusion joining EWS exon 7 to FLI-1 exon 6. Optimal conditions for amplifying the 574 bp fragment were as follow: 3 mm MgCl2 and 50 cycles of PCR consisting of denaturation 95° C for 15 seconds, annealing at 66° C for 1 minute, and elongation at 72° C for 1 minute 30 seconds. We then determined that these primers, the probe, and the PCR conditions enabled the detection of the presently reported EWS-FLI-1 fusions. Similarly, for the detection of other EWS-ETS fusions observed in Ewing tumors, the same EWS 3 tqm primer was used together with 3′ oligos corresponding to either ERG, ETV1, E1AF, or FEV genes. We verified that each individual fusion, except EWS-E1AF, for which no tumor material was available, could be reliably detected with these primers and that a mix of the five 3′ primers could be used for a multiplex analysis of these fusions (Fig. 1, Multiplex PCR II). The sensitivity of this multiplex detection will be described below.

Detection of specific gene fusions using multiplex fluorescent analysis of chromosome translocations (MFACT). Tumors harboring eight different fusions (1, EWS[ex7]-ERG[ex6]; 2, EWS[ex7]-ETV1[ex9]; 3, EWS[ex7]-FLI-1[ex8]; 4, EWS[ex10]-FLI-1[ex5]; 5, EWS[ex7]-FLI-1[ex6]; 6, PAX3-FKHR; 7, EWS-WT1; 8, SYT-SSX1]) were tested with both a conventional reverse transcription polymerase chain reaction (RT-PCR) approach (A) and a multiplex fluorescent analysis of chromosome translocations (MFACT) (B). A, The eight aforementioned tumors were analyzed by RT-PCR with specific primers. The top panel shows the EWS control amplification. The middle panel corresponds to the detection of the specific EWS-ETS fusions. The bottom panel shows the detection of specific PAX3-FKHR, EWS-WT1, and SYT-SSX. B, The RNAs used for the experiment shown in A were used for MFACT. PCR I is the control amplification with EWS-specific primers and an EWS-specific probe labeled with FAM. PCR II is aimed at the detection of Ewing-specific fusions with the same EWS probe. PCR III is a multiplex PCR used to detect the three indicated fusions with specific probes labeled with three different dyes.
For other gene fusions listed in Table 1, primers and Taqman probes were designed to be compatible with PCR conditions determined for EWS-ETS fusions of Ewing tumor apart from the MgCl2 concentration, which was adapted for an optimal detection of each fusion (Tables 1, 2, and 3). Primer pairs and probes were tested on tumor RNA previously demonstrated to exhibit the fusion of interest, except for SYT-SSX4, TFG-ALK, and ATIC-ALK, for which no control RNAs were available. Concerning synovial sarcoma (SS), the SSXc.3 tqm primer matches perfectly with the SSX1 sequence and exhibits the same mismatch with SSX2 and SSX4 sequences at position 4. We checked that the presence of this mismatch did not impair the detection of an SYT-SSX2 fusion. Although it was not tested, we anticipate that an efficient detection of SYT-SSX4 fusion would also be achieved with these conditions. For ARMS, multiplex analysis with primers Pax3.1 tqm, Pax7.1 tqm, and FKHR1.2 tqm was shown to be as efficient as single PCR in detecting PAX3-FKHR and PAX7-FKHR. Similarly, multiplex analyses of the fusions of extraskeletal myxoid chondrosarcoma, SS, and anaplastic lymphoma were validated.
Multiplex Fluorescent Analysis of Chromosome Translocations
To set up a diagnostic assay that could detect the most frequent gene fusions observed in small round cell tumors and sarcomas, we took advantage of the use of both multiplex analyses and different dyes for Taqman probes. The following assay consisting of three parallel PCRs was designed: the first PCR consisted of a control amplification of the ubiquitously expressed EWS gene and therefore evaluates the quality of the RNA; the second PCR is a multiplex analysis of the different Ewing-specific fusions as described above; the third PCR is a mix of primers and probes for detecting alveolar rhabdomyosarcoma (ARMS), SS, and small round cell desmoplastic tumors (SRCDT). For this last PCR, a specific labeling was used for each probe (Table 1).
The sensitivity of the multiplex detection of EWS-ETS fusions of Ewing tumors was determined using serial dilutions of control RNAs. As expected, a linear variation of the Ct (number of the cycle at the threshold) depending on the log of the amount of RNA was observed. The results observed for the control EWS amplification and three different EWS-FLI-1 fusions are shown in Figure 2. For the EWS transcript, a Ct lower than 40 was consistently observed for 10 pg of RNA (the estimated amount of RNA of a single cell). The sensitivities of detection for EWS-FLI-1 type 1 and type 2 (junction between EWS exon 7 and FLI-1 exon 5) were similar and slightly lower than that of the EWS control (Fig. 2). For the larger 892 bp fragment corresponding to a junction between EWS exon 10 and FLI-1 exon 5, a consistent detection of the PCR product with a Ct lower than 40 was observed for 1 ng of RNA. For amounts of RNA lower than 100 pg, this fusion could not be detected (Fig. 2). This result indicated that, as expected, the efficiency of the PCR decreases with the size of the amplification product.

Sensitivity of the detection of EWS-FLI-1 fusions using MFACT. The MFACT approach was used to analyze both EWS and EWS-FLI-1 fusion transcripts on serial dilutions of tumor RNA with a type 1, a type 2, or a type X fusion (between EWS ex 10 and FLI-1 ex 5). The amount of RNA (in picograms) is indicated on the x axis and the cycle threshold (Ct) on the y axis. For each fusion, three independent measures of Ct were performed for 100 ng, 10 ng, 1 ng, 100pg, 50pg, 20pg, and 10pg of cellular RNA. Curves are drawn using the mean of these three values. From this diagram it can be concluded that a Ct of 30 for the EWS control corresponds to an analysis of less than 1 ng of cellular RNA (around 100 cells). For this Ct, all types of EWS-FLI-1 transcripts can be detected. If the Ct for EWS is higher than 30, large EWS-FLI-1 transcripts might escape detection. Therefore, a Ct for EWS less than 30 is requested to avoid potential false-negative results for large fusion transcripts.
For the multiplex III (EWS-WT1, SYT-SSX1 and 2, PAX3, or 7-FKHR) and for all other fusions listed in Table 1, the sensitivities of detection were similar to that of the EWS control, ie, the fusions were always detected for 10 pg of RNA with a Ct lower than 40. Therefore, the detection of large EWS-FLI-1 transcripts represents the limit of application of this technique: at least 1 ng of RNAs (around 100 cells) should be used to avoid false-negative results for EWS-FLI-1 fusions. This amount corresponds to a Ct for the EWS control lower than 30 (Fig. 2).
Validation of This Assay on a Series of 79 Tumors
Seventy-nine tumors, referred to the laboratory for the study of gene fusions, were analyzed in parallel by multiplex fluorescent analysis of chromosome translocations (MFACT) and conventional PCR. Results are shown in Table 4. Six cases were assumed noninterpretable (NI) by conventional RT-PCR because the EWS control fragment could not be observed by ethidium bromide staining of agarose gel (Delattre et al, 1994). These six cases demonstrated a Ct for EWS higher than 30 by MFACT. One additional case exhibited a Ct of 30.7 by MFACT and was thus considered as NI by this approach. Thirty-nine cases were positive for EWS-ETS fusions by MFACT, compared with 38 by conventional RT-PCR. The discordant case involved an Ewing tumor confirmed by pathological examination; however, the fragment of tumor received by the laboratory contained only a few tumor cells. Accordingly, the Ct for EWS-ETS fusion was 42, whereas that of the EWS control was 28. Nine tumors were positive for PAX 3 or 7-FKHR by MFACT, compared with 8 by conventional RT-PCR. Interestingly, the discordant case here was a bone marrow aspirate containing a small number of tumor cells from a pathologically confirmed ARMS. Three cases were positive by both approaches for SS and SRCDT. Altogether, 21 cases were negative by conventional RT-PCR and 18 by MFACT. These results indicate that both approaches yield highly consistent results. However, MFACT appears slightly more efficient because two fusions ignored by conventional RT-PCR could be detected by MFACT. Both cases involved samples containing a small number of tumor cells.
Discussion
We describe primers, probes, and PCR conditions that enable one to detect efficiently most gene fusions observed in solid tumors. The standardization of the reverse transcriptase and PCR cycling conditions enable one to search for these different fusions on the same plate during a single round of PCR. Furthermore, the use of different dyes for the labeling of probes permit multiplex PCR and detection. We propose an assay, termed MFACT, that detects the most frequent fusions observed in sarcomas and small round cell tumors, including those of Ewing tumor, ARMS, SS, and SRCDT. As an internal control, the amplification of the ubiquitously expressed EWS gene is used. This control appears particularly appropriate because its level of expression is similar to those of the tested gene fusions.
This highly specific method considerably simplifies and reduces the bench work because all the post-PCR steps are suppressed. Consequently, no PCR products are manipulated, which considerably reduces the risk of cross-contamination. In addition, apart from the isolation of RNA, all other steps can follow a fully automated process.
We show that, except for large fusion transcripts of Ewing, the PCR conditions enable the systematic detection of all fusions when 10 pg of tumor RNA, the equivalent of one cell, are used. Taking into account the lower sensitivity for the detection of Ewing transcripts, we propose that a Ct for the EWS control less than 30 should be observed for a fully reliable interpretation of results.
Since the set up of this approach in the lab, 799 tumors have been analyzed by MFACT. One hundred one cases (13%) were considered noninterpretable, given a Ct for EWS greater than 30. The usual causes were the very small size of the tumor fragments, the necrosis of the fragment, or the poor condition of the sampling. Three hundred fourteen tumors presented specific transcripts as follows: EWS-ETS in 215, SYT-SSX in 43, PAX-FKHR in 35, and EWS-WT1 in 21 cases. A subset of the 799 tumors was analyzed for specific fusions not included in the MFACT assay and revealed 4 EWS-ATF1, 2 EWS/TAFII68-TEC, 5 anaplastic lymphoma fusions, and 2 ETV6-NTRK3 fusions. Finally, 371 tumors were negative for the tested fusions. Interestingly, this analysis shows that no tumor exhibits two different fusions, which confirms the association of these fusions with specific tumor types and which strongly suggests the absence of false-positive cases, thus reinforcing the reliability of the test.
In SS (Inagaki et al, 2000; Kawai et al, 1998; Nilsson et al, 1999), ARMS (Kelly et al, 1997), and Ewing tumor (de Alava et al, 1998; Zoubek et al, 1994), the types of fusion transcripts have been associated with prognostic information suggesting that a precise typing of fusion transcripts might be of clinical interest. By itself, the presently described method does not enable a precise typing of these transcripts. Currently, to type fusion transcripts, we perform a standard PCR with gel electrophoresis and Southern hybridization with specific probes. The primers used for typing are internal to those used for the MFACT. Therefore, the typing step cannot lead to a contamination of the diagnostic step. Alternatively, the real-time PCR could be used for a precise typing of fusion transcripts using pairs of primers and probes specific for certain types of fusion.
In conclusion, the real-time PCR constitutes a simple and efficient method for the detection of the gene fusions observed in human solid tumors. Although this was not tested in the present study, the sensitivity of this approach should enable its use for the detection of residual and minimal disease.
Materials and Methods
RNA Isolation, Reverse Transcriptase
Tumor samples were snap-frozen in liquid nitrogen. RNA was isolated using the Trizole extraction kit (Gibco BRL, Gaithersburg, Maryland). A total of 1 μg of total RNA was reverse transcribed using random hexamers in a final volume of 20 μl using the GeneAmp RNA PCR Kit (PE Biosystems, Foster City, California).
Real-Time PCR
Real-time PCR experiments were performed in a final volume of 50 μl containing 2 μl of cDNA, with 200μm each of dATP, dCTP, and dGTP; 400μm of dUTP; 200 nm of each primer; 100 nm of the Taqman probe; 1.5 U AmpliTaq Gold (PE Biosystems); and 0.5 U AmpErase UNG (Uracile N Glycosylase, PE Biosystems). After initial steps of UNG reaction for 2 minutes at 50° C and TaqGold activation for 15 minutes at 95° C, 50 cycles of PCR were performed according to standardized procedures (denaturation at 95° C for 15 seconds, annealing at 66° C for 1 minute, and elongation at 72° C for 1.5 minutes). The primers and probes are described in Tables 2 and 3, respectively. The only variable parameter was the MgCl2 concentration. Optimal concentrations of MgCl2 for the detection of individual fusions are indicated in Table 1. For the MFACT test, the multiplex detection of PAX-FKHR, SYT-SSX, and EWS-WT1 was performed at a concentration of 4 mm of MgCl2.
The real-time PCR was carried out using the ABI/PRISM 7700 (PE Applied Biosystems). The fluorescence data were collected during the annealing and extension phases of every cycle.
Conventional PCR Conditions
Standard PCR was performed using the GeneAmp PCR Core Reagents kit N808-0009 (PE Biosystems) in a reaction mixture of 25 μl containing 2 μl of cDNA, 200μm of each dNTP, 500 nm of each primer, 1.5 mm of MgCl2, and 0.7 U TaqDNA polymerase. The sequence of primers used for conventional PCR can be obtained upon request.
References
Attwooll C, Tariq M, Harris M, Coyne JD, Telford N, and Varley JM (1999). Identification of a novel fusion gene involving hTAFII68 and CHN from a t(9;17)(q22;q11.2) translocation in an extraskeletal myxoid chondrosarcoma. Oncogene 18: 7599–7601
Barr FG (1998). Translocations, cancer and the puzzle of specificity. Nat Genet 19: 121–124.
Bennicelli JL and Barr FG (1999). Genetics and the biologic basis of sarcomas. Curr Opin Oncol 11: 267–274.
Clark J, Benjamin H, Gill S, Sidhar S, Goodwin G, Crew J, Gusterson BA, Shipley J, and Cooper CS (1996). Fusion of the EWS gene to CHN, a member of the steroid/thyroid receptor gene superfamily, in a human myxoid chondrosarcoma. Oncogene 12: 229–235.
Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, Gusterson BA, and Cooper CS (1994). Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 7: 502–508.
Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A, Shipley J, Gusterson BA, and Cooper CS (1995). Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J 14: 2333–2340.
Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, and Barr FG (1994). Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 54: 2869–2872.
de Alava E, Kawai A, Healey JH, Fligman I, Meyers PA, Huvos AG, Gerald WL, Jhanwar SC, Argani P, Antonescu CR, Pardo-Mindan FJ, Ginsberg J, Womer R, Lawlor ER, Wunder J, Andrulis I, Sorensen PH, Barr FG, and Ladanyi M (1998). EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol 16: 1248–1255.
Delattre O, Zucman J, Melot T, Sastre X, Zucker JM, Lenoir G, Ambros P, Sheer D, Turc-Carel C, Triche T, Aurias A, and Thomas G (1994). The Ewing family of tumors: A subgroup of small round cell tumors defined by specific chimeric transcripts. New Engl J Med 331: 294–299.
Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, and Thomas G (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumors. Nature 359: 162–165.
de Leeuw B, Balemans M, Olde Weghuis D, and Geurts van Kessel A (1995). Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum Mol Genet 4: 1097–1099.
Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, Emanuel BS, Rovera G, and Barr FG (1993). Fusion of a fork head domain gene to PAX3 in the solid tumor alveolar rhabdomyosarcoma. Nat Genet 5: 230–235.
Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A, Lamant L, Falini B, Ott G, Mason DY, Delsol G, and Campo E (1999). TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 94: 3265–3268.
Inagaki H, Nagasaka T, Otsuka T, Sugiura E, Nakashima N, and Eimoto T (2000). Association of SYT-SSX fusion types with proliferative activity and prognosis in synovial sarcoma. Mod Pathol 13: 482–488.
Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, and Shapiro DN (1995). A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 10: 1229–1234.
Kaneko Y, Yoshida K, Handa M, Toyoda Y, Nishihira H, Tanaka Y, Sasaki Y, Ishida S, Higashino F, and Fujinaga K (1996). Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes Chromosomes Cancer 15: 115–121.
Kawai A, Woodruff J, Healey JH, Brennan MF, Antonescu CR, and Ladanyi M (1998). SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med 338: 153–160.
Kelly KM, Womer RB, Sorensen PH, Xiong QB, and Barr FG (1997). Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol 15: 1831–1836.
Knezevich SR, McFadden DE, Tao W, Lim JF, and Sorensen PH (1998). A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 18: 184–187.
Labelle Y, Zucman J, Stenman G, Kindblom LG, Knight J, Turc-Carel C, Dockhorn-Dworniczak B, Mandahl N, Desmaze C, Peter M, Aurias A, Delattre O, and Thomas G (1995). Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum Mol Genet 4: 2219–2226.
Ladanyi M and Bridge JA (2000). Contribution of molecular genetic data to the classification of sarcomas. Hum Pathol 31: 532–538.
Ladanyi M and Gerald W (1994). Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor. Cancer Res 54: 2837–2840.
Lamant L, Dastugue N, Pulford K, Delsol G, and Mariame B (1999). A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 93: 3088–3095.
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, and Look AT (1994). Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263: 1281–1284.
Nilsson G, Skytting B, Xie Y, Brodin B, Perfekt R, Mandahl N, Lundeberg J, Uhlen M, and Larsson O (1999). The SYT-SSX1 variant of synovial sarcoma is associated with a high rate of tumor cell proliferation and poor clinical outcome. Cancer Res 59: 3180–3184.
Panagopoulos I, Mencinger M, Dietrich CU, Bjerkehagen B, Saeter G, Mertens F, Mandahl N, and Heim S (1999). Fusion of the RBP56 and CHN genes in extraskeletal myxoid chondrosarcomas with translocation t(9;17)(q22;q11). Oncogene 18: 7594–7598.
Peter M, Couturier J, Pacquement H, Michon J, Thomas G, Magdelenat H, and Delattre O (1997). A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 14: 1159–1164.
Peter M, Mugneret F, Aurias A, Thomas G, Magdelenat H, and Delattre O (1996). An EWS/ERG fusion with a truncated N-terminal domain of EWS in a Ewing’s tumor. Int J Cancer 67: 339–342.
Rosenwald A, Ott G, Pulford K, Katzenberger T, Kuhl J, Kalla J, Ott MM, Mason DY, and Muller-Hermelink HK (1999). t(1;2)(q21;p23) and t(2;3)(p23;q21): Two novel variant translocations of the t(2;5)(p23;q35) in anaplastic large cell lymphoma. Blood 94: 362–364.
Shapiro DN, Sublett JE, Li B, Downing JR, and Naeve CW (1993). Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res 53: 5108–5112.
Sjogren H, Meis-Kindblom J, Kindblom LG, Aman P, and Stenman G (1999). Fusion of the EWS-related gene TAF2N to TEC in extraskeletal myxoid chondrosarcoma. Cancer Res 59: 5064–5067.
Skytting B, Nilsson G, Brodin B, Xie Y, Lundeberg J, Uhlen M, and Larsson O (1999). A novel fusion gene, SYT-SSX4, in synovial sarcoma. J Natl Cancer Inst 91: 974–975.
Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, and Denny CT (1994). A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 6: 146–151.
Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG, and Falini B (2000). A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res 60: 793–798.
Zoubek A, Pfleiderer C, Salzer-Kuntschik M, Amann G, Windhager R, Fink FM, Koscielniak E, Delattre O, Strehl S, Ambros PF, Gadner H, and Kovar H (1994). Variability of EWS chimaeric transcripts in Ewing tumours: A comparison of clinical and molecular data. Br J Cancer 70: 908–913.
Zucman J, Delattre O, Desmaze C, Epstein AL, Stenman G, Speleman F, Fletchers CD, Aurias A, and Thomas G (1993a). EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 4: 341–345.
Zucman J, Melot T, Desmaze C, Ghysdael J, Plougastel B, Peter M, Zucker JM, Triche TJ, Sheer D, Turc-Carel C, Ambros P, Combaret V, Lenoir G, Aurias A, Thomas G, and Delattre O (1993b). Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J 12: 4481–4487.
Acknowledgements
We thank Henri Magdelenat for help in the Taqman analysis and the following colleagues: Valérie Com- baret, Jean Michon, Odile Oberlin, Thierry Phillip, and Marie-José Terrier-Lacombe, for providing tumor samples.
This work was supported by grants from the Programme Hospitalier de Recherche Clinique and the Institut Curie.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peter, M., Gilbert, E. & Delattre, O. A Multiplex Real-Time PCR Assay for the Detection of Gene Fusions Observed in Solid Tumors. Lab Invest 81, 905–912 (2001). https://doi.org/10.1038/labinvest.3780299
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780299
This article is cited by
-
Establishment of multiplex RT-PCR to detect fusion genes for the diagnosis of Ewing sarcoma
Diagnostic Pathology (2021)
-
Alveolar soft part sarcoma—radiologic patterns in children and adolescents
Pediatric Radiology (2013)
-
A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion
Nature Genetics (2012)
