
Abstract
Hyperplastic glomerular epithelial lesion is an important determinant of the progression of idiopathic focal segmental glomerulosclerosis (FGS). The proliferation and differentiation of glomerular epithelial cells and parietal epithelial cells (PECs) are regulated differently by cyclin and cyclin-dependent kinase inhibitors (CKIs) during nephrogenesis. To access the cellular mechanism underlying epithelial hyperplasia in the development of FGS, the present study applied immunohistochemistry to 21 cases of FGS to demonstrate expression of cell-cycle molecules and phenotypic characterization in proliferative epithelial lesions in FGS. The materials included segmental sclerosis (18.1%), which was divided into monolayer epithelial lesions (64.6%) and cellular lesions (35.4%). All of the cellular lesions expressed cytokeratin, frequently with Ki-67 (82.4%) and less frequently with cyclin A (17.7%), but were invariably negative for podocyte markers (PHM-5 and synaptopodin) and CKIs (p27kip1 and p57kip2). Podocytes in nonsclerotic tuft in the same glomeruli with cellular lesions strongly expressed CKIs and podocyte markers. Moreover, electron microscopy showed that some large proliferating cells with prominent nucleoli have a broad cell base attached to Bowman’s capsule. These cells have cilia and a junctional complex with neighboring hyperplastic cells, some of which directly cover the glomerular basement membrane. This suggests that cellular lesions are of PEC origin. Monolayer epithelial lesions also exclusively exhibited a PEC phenotype with reciprocal expression of podocyte markers and cytokeratin. In addition, CKIs are weakly expressed in monolayer epithelial lesions, suggesting a re-entry of cell-cycle quiescent. In conclusion, proliferation of PEC, sustained by repression of CKIs in nature and simultaneous activation of cyclin A, is the actual molecular background to the cellular lesions in FGS. Cellular lesions may result in monolayer epithelial lesions that retain the PEC phenotype and enter a common pathway to glomerulosclerosis.
Similar content being viewed by others


Introduction
Idiopathic focal segmental glomerulosclerosis (FGS) is a primary renal disease frequently associated with steroid-resistant nephrotic syndrome. Despite various therapeutic strategies to prevent disease progression, the majority of cases of FGS progress to chronic renal failure (Cameron, 1996). Although frequent recurrence of FGS after renal transplantation suggests involvement of humoral factors in the pathogenesis of this disease (Dantal et al, 1994; Savin et al, 1996), the mechanism by which the glomeruli undergo sclerosis is still obscure. The pathological findings in FGS have revealed a nonspecific pattern of glomerulosclerosis including glomerular capillary collapse concurrent with deposition of extracellular matrix or hyalinosis (D’Agati, 1994). Earlier studies that relied on electron microscopy have shown that podocyte degeneration and detachment from the glomerular basement membrane (GBM) are the initial histological events in glomerulosclerosis in FGS (Grishman and Churg, 1975).
In addition to podocyte degeneration, epithelial hyperplasia within the glomeruli has been suggested as an important determinant of the progression of FGS (Schwartz and Lewis, 1985; Schwartz et al, 1999). This condition is commonly referred to as “cellular lesion” and is characterized by two or more layers of hypertrophic and hyperplastic epithelial cells overlying sclerotic or collapsed tuft segments. Patients with cellular lesions have an increased prevalence of end-stage renal disease compared with FGS patients without cellular lesions (Schwartz et al, 1999). In previous reports, the terms indicating the type of cells involved in cellular lesions varied from author to author. Schwartz and Lewis simply described them as “glomerular epithelial cells” (Schwartz and Lewis, 1985), and other authors have called them “visceral epithelial cells” (Grishman and Churg, 1975; D’Agati, 1994). Churg and Sorbin refer to the lesions as “podocyte hyperplasia” (Churg and Sorbin, 1982). These terms are based on their localization related to the GBM, and are not based on phenotypic features, which are particularly important in providing further clues to understanding the mechanism responsible for the development of sclerosis in FGS. In addition, investigation of the cell-cycle molecules expressed in the lesions may provide an unique insight into the pathogenesis of FGS, because the quiescent cell cycle in podocytes (Pabst and Sterzel, 1983) and the cell-cycle activation in hyperplastic epithelial cells may play key roles in the pathogenesis of FGS.
The cell cycle is ultimately regulated by the balance between positive and negative cell-cycle molecules (Shankland, 1997a [Review]). Cyclin-dependent kinase inhibitors (CKIs), a negative regulator of the cell cycle that determines cell-cycle arrest, have been found to be distributed in quiescent cells, and their level declines when the cells are stimulated to proliferate (Polyak et al, 1994). Recently, we and others have found that podocytes specifically express CKIs, namely, p27Kip1 and p57Kip2, concurrent with loss of cell-proliferating marker and cyclin expression in human fetal kidneys (Combs et al, 1998; Nagata et al, 1998b). This cell-cycle stability in podocytes correlated closely with an expression of specific differentiation markers. Because mature podocytes were absolutely negative for proliferating markers, such as Ki-67 and proliferating cell nuclear antigen after nephrogenesis, and they continued to express CKIs thereafter (Combs et al, 1998; Nagata et al, 1993; Nagata et al, 1998b), we speculate that CKIs play an active role in cell-cycle stability and terminal differentiation in podocytes.
To access the molecular background of epithelial lesions in FGS, the present study used immunohistochemistry to demonstrate expression of cell-cycle molecules and podocyte differentiation markers in renal biopsy specimens from idiopathic FGS.
Our results indicate that proliferation of parietal epithelial cells (PECs), sustained by repression of CKIs in nature, is the actual molecular background of cellular lesions in FGS. Cellular lesions may result in monolayer epithelial lesions with synechiae, which is a gate to the common pathway for progressive glomeulosclerosis.
Results
Background Histology
Figure 1 shows representative features of the epithelial pathology in the glomeruli observed in our material. The epithelial lesions can be classified as either monolayer or cellular lesions. Endocapillary or mesangial proliferation was frequently found in the tuft beneath cellular lesions but was generally absent in monolayer lesions. In total, 531 glomeruli from 21 patients were examined by single-section analysis and 96 of them (18.1%) were found to show segmental glomerulosclerosis or tuft collapse with epithelial lining. Sixty-two of them exhibited monolayer epithelial cells covering the sclerosis (64.6%), and 34 glomeruli showed “cellular lesions” (35.4%). Monolayer lesions were usually associated with synechiae between the tuft and Bowman’s capsule, and cellular lesions were also frequently found to be transposed to cuboidal PECs on serial section analysis.

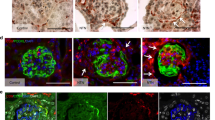
Epithelial features in FGS. Various features of the epithelial lesions in FGS demonstrated by PAM staining. A, small focus of epithelial aggregation at the site of segmental vascular pole sclerosis (arrowhead). B, A monolayer of epithelial cells with synechiae covers a sclerotic tuft segment. C to E, Cellular lesions. Note that epithelial hyperplasia covers sclerotic and collapsed tuft segments. F, High magnification of E. Cytoplasmic droplets, many vacuoles, and bleb formation, which are consistent with the structure of damaged podocytes, are observed at the periphery of the hyperplastic epithelial cells (arrows). Note, the adjacent parietal epithelial cells (PECs) are hyperplastic. (A to C, × 200; D, × 320; E, × 180; F, × 430)
Immunoreaction in Control Kidneys
Figure 2 exhibits podocyte markers, cytokeratin, and cell-cycle protein expression in control kidneys. PHM-5 and synaptopodin were expressed exclusively in podocytes, but never in PEC. Cytokeratin was very frequently (approximately 80%) expressed in the PEC and but not at all in the podocytes. Cuboidal PECs near the urinary pole tended to be more frequently cytokeratin-positive than flat PECs located near the vascular pole. p27Kip1 and p57Kip2 were exclusively expressed in podocyte nuclei, but never in PECs. Cyclin A was absent from podocytes and PECs. CD 68 and Ki-67 were not expressed in podocytes. Ki-67 was sometimes detected in PEC (approximately 10%).

Expression of cell-cycle proteins and epithelial markers in control kidneys. The localization of PHM-5 and synaptopodin is restricted to podocytes. Cytokeratin is expressed in a considerable number of PEC, but not at all in podocytes. Flat PECs near the vascular pole are negative for cytokeratin. p27Kip1 and p57Kip2 are expressed only in podocytes. Cyclin A is absent in podocytes and PECs, but slightly expressed in tubular cells (arrowheads). (Cytokeratin and cyclin A staining, × 130; others, × 160)
Both Types of Epithelial Lesions Express Cytokeratin, But Not Podocyte Markers
Serial section analysis demonstrated that all of the cells in monolayer lesions covering the sclerotic tuft segment expressed cytokeratin but were negative for podocyte markers. The reciprocal expression pattern of cytokeratin and the podocyte markers was particularly noteworthy (Fig. 3, B and C). Likewise, almost all of the cells in all of the cellular lesions showed cytokeratin expression of variable intensity (Fig. 3F; Fig. 4F). Synaptopodin and PHM-5 expression coincided precisely (Fig. 3, G and H), and both were entirely absent from the foci with cellular lesions (Fig. 3E; Fig. 4I). Cytokeratin-positive epithelial lesions frequently had a connection with PECs, which also expressed cytokeratin. CD 68-positive cells were mostly observed within glomerular capillary, if present, but not observed in any feature of cellular lesions (Fig. 3I).

Phenotypic characterization of epithelial lesions. A to C are serial sections of monolayer lesions. Cytokeratin-expressing tuft segments are negative for PHM-5 and vice versa (reciprocal expression, arrows). D to F are serial sections of cellular lesions (arrows). Cellular lesions expressed cytokeratin, but not PHM-5. Note cellular lesions transpose to PECs which also express cytokeratin. G and H are serial sections of cellular lesions. The expression patterns of two markers of podocytes, synaptopodin (G) and PHM-5 (H), are quite similar. CD 68-positive cells (I) are found within the capillaries, but not in the cellular lesions. Ki-67 (J) is frequently expressed among hyperplastic cells in cellular lesions. (A to C, × 100; D and E, × 180; F, × 450; G to J, × 140)

Expression of CKIs and phenotypic markers in epithelial lesions. A to C are serial sections of monolayer epithelial lesions. P27Kip1 and p57Kip2 are occasionally positive in monolayer epithelial cells (arrowheads) which express cytokeratin. Note the different staining patterns for p27Kip1 and p57Kip2. D to I are serial sections of cellular lesions. The expression patterns of p27 Kip1 and p57 Kip2 in cellular lesions are identical and restricted to podocytes. p27 Kip1 and p57 Kip2 are not expressed in cellular lesions (arrowheads), but intensely expressed in podocytes of nonsclerotic tuft segments. These CKI-negative cellular lesions expressed cytokeratin (F), but not synaptopodin (I). J to L are serial sections of an epithelial lesion resembling a cellular crescent. Hyperplastic PECs are negative for p27Kip1 (J), but are frequently positive for Ki-67 (K) and cytokeratin (L). Note, podocytes in preserved glomerular tuft express p27Kip1 (J). (A to C, × 330; D to L, × 150)
Cellular Lesions Lacked p27Kip1 and p57Kip2 Expression
As shown in Figure 4, monolayer lesions and transposed PEC expressed somewhat weak, but distinct, immunoreactivity for p27Kip1, whereas p57kip2 in the same lesion was expressed less often (Fig. 4, A, B, and C). In cellular lesions, neither of these two CKIs was expressed. The repressed expression of CKIs in cellular lesions was enhanced by the intense immunoreactivity in podocytes in nonsclerotic glomerular tuft in the same glomeruli (Fig. 4, D, E, G, and H). CKI-repressed cellular lesions frequently expressed cytokeratin. Ki-67 expression was seen in 82.4% of cellular lesions (Fig. 3J) but seldom in monolayer lesions. As shown in Figure 4, J, K, and L, the hyperplastic PECs that formed cellular crescents frequently expressed Ki-67 and cytokeratin but were negative for CKIs. Cyclin A-positive cells were found in 17.7% of cellular lesions. Cyclin A-expressing cells were found against the background of CKI-negative cellular lesions (Fig. 5). Monolayer epithelial lesions did not contain cyclin A-positive cells.

Representative cellular lesions stained with p27 Kip1 and cyclin A. Two representative glomeruli with serial sections. p27Kip1 negative cellular lesions contain some cyclin A positive cells. Note the cyclin A positive cells in the PECs (B, arrowheads) and in the cellular lesions (D, arrowhead). (A and B, × 180; C and D, × 200)
Loss of CKI Expression in Podocytes of Nonsclerotic Glomeruli
Although scarce (4.6% of all nonsclerotic glomeruli), CKI-negative podocytes were found in nonsclerotic glomeruli in FGS, and this was not seen in control kidneys (Fig. 6). The nuclei of these CKI-negative podocytes were somewhat larger than those in surrounding CKI-expressing podocytes. Phenotypic alteration to CKI-negative podocytes could not be assessed because of technical limitations in the serial sectioning for single cells. Results of immunohistochemistry are summarized in Table 1.

Lack of CKI expression in podocytes of nonsclerotic glomeruli in FGS. Representative glomeruli reveal p57 Kip2 (A and B) and p27 Kip1 (C) negative podocytes in nonsclerotic glomeruli (arrowheads). As shown in B, nuclei of some CKI-negative podocytes are relatively large (arrowheads) and the expression of CKIs in surrounding podocytes is distinct. (A to C, × 400)
Ultrastructure of Cellular Lesions Reveals Characteristic PEC Morphology
Electron micrographs of cellular lesions (Fig. 7) showed that hyperplastic epithelial cells with prominent nucleoli are frequently located on the parietal side, and some have a cellular base on the Bowman’s capsule. Each hyperplastic cell has a connection with a junctional complex, and some of the cells directly cover the GBM without having actin bundles at the cell base (Fig. 8). In close proximity to GBM, we found atrophic or apoptotic cells with cytoplasmic vacuoli and protein droplets that resemble damaged podocytes. In the preserved tuft segment of the glomeruli with cellular lesions, binucleated podocytes were sometimes observed. Underlying glomerular tufts of cellular lesions are collapsed and have foamy macrophages.

Electron micrograph of a cellular lesion. Three layers of epithelial cells are shown. Large cells with prominent nucleoli are predominantly located on the parietal side. Some of them have a broad cell base attached to Bowman’s capsule. Note the pyknotic cells and degenerated cells (arrow) near the glomerular basement membrane (× 3000).

Electron micrographs of a monolayer lesion. Large cells with prominent nucleoli have junctional complexes with parietal epithelial cells (arrowheads) and neighboring cells (thick arrows), the latter directly underlying the glomerular tuft. Note the absence of cytoplasmic actin bundles (thin arrows). One of these cells has cilia (open arrow)(× 5700). Inset: Greater magnification of the adherence junction (thick arrow) (× 40000).
Discussion
The present study showed cell-cycle protein expression in FGS. Candidates for the cellular components of cellular lesions are podocytes and PEC, the latter including transposed proximal tubular epithelial cells. These cells are of the same lineage: from the metanephric mesenchyme to early S-shaped body stage of nephrogenesis. We have recently demonstrated that up-regulation of CKIs and the differentiation marker coincided precisely with down-regulation of the proliferating marker and cell-cycle promoters, cyclin A and cyclin B1, in podocytes in human fetal kidneys. By contrast, PECs do not express CKIs and continue to express cytokeratin after the capillary-loop stage, suggesting that the cell-cycle regulatory mechanism involves phenotypic diversion in both epithelial cells (Moll et al, 1991; Nagata et al, 1998b). We have surmised that the cellular events occurring in the proliferating cellular lesions in glomerular disease mimic the dramatic cellular changes to podocytes and PEC during nephrogenesis, and thus phenotypical characteristics and cell-cycle molecules of cellular lesions may provide novel clues for understanding the molecular background of the pathogenesis of FGS.
The present study provides evidence that cellular lesions have Ki-67- and cyclin A-positive cells, but are entirely negative for two CKIs, namely p27Kip1 and p57Kip2. This suggests that repression of CKIs and activation of cyclins is an actual molecular background for cellular lesions. There are two possible explanations for CKI repression in cellular lesions. One is the proliferation of PECs, which have low levels of CKIs in nature, and the other is dysregulation of CKIs in podocytes. In this regard, the cellular origin of the lesions is particularly important to understanding the molecular mechanisms responsible for FGS.
Based on several lines of evidence, we suggest that cellular lesions are of PEC origin. First, podocytes are considered post-mitotic cells and may undergo nuclear division without cytokinesis in renal disease or on exposure to a strong mitogen, such as basic-fibroblast growth factor (Kriz et al, 1995; Nagata et al, 1995b). Cytokinesis in mature podocytes has not been established in vivo. Second, post-mitotic characteristics in podocytes are associated with a strong expression of CKIs, and up-regulation of CKIs limits podocyte proliferation in experimental membranous glomerulonephritis (Shankland et al, 1997b), suggesting that proliferation of podocytes is tightly restricted by the CKI regulatory mechanism. This is supported by the current finding that podocytes in the nonsclerotic tuft segment within the same glomeruli having cellular lesions strongly expressed CKIs. Third, the present study extended the previous notion that cellular lesions usually have a connection to PECs, and the ultrastructure of cellular lesions exhibited several characteristics of PECs, but not podocytes (Kihara et al, 1997). In contrast to podocytes, mature PECs in normal glomeruli sometimes express Ki-67 and are virtually negative for CKIs. Furthermore, a typical example of PEC proliferation, usually referred to as cellular crescents, is frequently found in various renal diseases. This suggests that PECs potentially proliferate in response to various kinds of stimuli. Moreover, the present study clearly demonstrated that cellular lesions are virtually cytokeratin-positive and -negative for synaptopodin and PHM-5. These findings strongly support our notion that the majority of cellular lesions are of PEC origin. Thus, it is likely that proliferation of CKI-repressing PECs in nature is an actual mechanism of cellular lesions in FGS.
Several reports have suggested podocyte proliferation in collapsing glomerulopathy or FGS from the immunohistochemical findings of so-called phenotypic changes. Loss of podocyte markers, namely, synaptopodin or GLEPP-1 in collapsing glomerulopathy or FGS, has been recently reported (Barisoni et al, 1999; Sharif et al, 1998). Because loss of cell markers is not equal to phenotypic changes, it may be that a decreased expression of podocyte markers simply indicates a failure of protein synthesis caused by cell injury, thus resulting in insufficient proliferation. On the other hand, (Bariety et al 1998) demonstrated expression of macrophage markers in podocytes. We previously found cytokeratin and podocyte marker double-positive cells in collapsing glomerulopathy (Nagata et al, 1998a). These findings indicate that phenotypic changes in podocytes occur in the FGS or collapsing glomerulopathy. Because cells with phenotypic changes are not necessarily dividing, a novel expression of some proteins in podocytes does not indicate proliferation. Together, there is no direct in vivo evidence for the proliferation of mature podocytes. One possible explanation supporting the hypothesis that cellular lesions are of podocyte origin is that proliferating presumptive podocytes express cytokeratin and cyclin A and are negative for CKIs and podocyte markers. However, this possibility is unlikely, because electron micrographs of cellular lesions in this study showed enlarged epithelial cells having prominent nucleoli predominantly located on the parietal side. These cells frequently exhibit a broad cell base attached to Bowman’s capsule and have connections with neighboring cells via junctional complexes (Fig. 8). Moreover, some of these hyperplastic cells directly cover the GBM without including the cytoplasmic actin bundles that are present in podocytes. Finally, degenerated cells, including protein droplets and swollen lysosomes, which are characteristic of damaged podocytes in FGS (Grishman and Churg, 1975), are sporadically found in close proximity to underlying GBM. Interestingly, we found CKI-negative podocytes in 4.6% of glomeruli without cellular lesion in FGS. In fact, we have previously noted that podocytes undergo mitosis and show binucleated cells in FGS. Thus the existence of CKI-negative podocytes would imply that podocytes potentially undergo M phase nuclear division, but this division is insufficient for cell proliferation. Recently, it has been shown that p21-deficient mice with Heyman nephritis demonstrated re-engagement of the cell cycle of podocytes with the marked proliferation that leads to sclerosis (Kim et al, 1999). This animal experiment would suggest podocyte proliferation caused by down-regulation of CKIs. Further studies are needed to investigate the regulatory mechanism of CKIs in podocytes and PECs and to determine whether podocytes undergo cytokinesis in vivo.
The sequence of events from cellular lesions to sclerosis is unknown. (Schwartz and Lewis 1985) have shown that cellular lesions were more prevalent in cases where the interval from the onset of proteinuria to renal biopsy was short compared with the cases of patients with segmental scars only. Moreover, repeat biopsies of FGS with cellular lesions in our previous study showed an increase of segmental scaring with monolayer epithelial lesions concurrent with a decrease of cellular lesions (Hattori et al, 1997). Thus, it may be that cellular lesions undergo segmental scaring with monolayer lesions. This notion is supported by the present study’s conclusion that both cellular lesions and monolayer lesions are the same phenotype of PECs. Of note, monolayer lesions with the PEC phenotype expressed CKIs somewhat weakly, but with a distinct immunoreactivity. Recently it has been shown that p27kip1 protected mesangial cells and fibroblasts from apoptosis (Hiromura et al, 1999). Thus, CKI-negative cellular lesions may reduce cell numbers via apoptosis, resulting in monolayer epithelial lesions, as found in the repair process of cellular crescents. This is also known as PEC hyperplasia (Shimizu et al, 1996). Up-regulation of CKIs in monolayer lesions may stop apoptosis to maintain epithelial cover for GBM.
Predisposing lesions of monolayer epithelial lesions in FGS are not restricted to cellular lesions. An early electron microscopic study suggested that podocyte detachment was the initial lesion prior to sclerosis (Grishman and Churg, 1975). Recent studies of various experimental models of glomerulosclerosis indicate that podocyte detachment and simultaneous compensation by PEC are a common pathway to sclerosis (Kriz et al, 1994). Because these epithelial features are frequently seen in various human renal diseases, including diabetic glomerulosclerosis and IgA glomerulonephritis (known as a mesangial disorder), the lesions seem to be nonspecific and the common pathologic alteration leading to glomerulosclerosis. Thus, we surmise that cellular lesions and podocyte detachment in FGS result in monolayer lesion of PECs and a nonspecific pathway to sclerosis as a tissue-repair mechanism. Because the characteristic behaviors of PECs are different from those of podocytes, PECs in monolayer lesions may disturb tuft reconstruction, ie, angiogenesis, in the collapsed tuft and accumulate abnormal extra-cellular matrices, thus leading to sclerosis.
In conclusion, proliferation of PECs, sustained by repression of CKIs in nature, is an actual molecular mechanism of cellular lesions in FGS. Cellular lesions may result in monolayer epithelial lesions retaining the PEC phenotype and entering the nonspecific pathway of glomerulosclerosis.
Materials and Methods
Biopsy Samples
Renal biopsy materials from 21 cases of FGS (male, 10; female, 11; average age at biopsy, 17.5 yrs; average proteinuria at biopsy, 5.8 g/day) were analyzed by histological examination and immunohistochemical staining. Eighteen cases were pediatric patients with steroid-resistant nephrotic syndrome, and ten patients developed chronic renal failure approximately 5 years after the onset of symptoms. The material in all cases included segmental sclerosis characterized by capillary collapse with simultaneous matrix deposition as defined by the International Study of Kidney Disease in Children (Churg et al, 1970). Immunofluorescence microscopy revealed segmental lumpy deposition of IgM and C3 in 11 cases at the sclerotic glomerular tuft. IgG, IgA, and C3 were not diffusely positive in any of the cases. All antibodies for immunofluorescence were purchased from ICN Pharmaceuticals, Inc. (Cleveland, Ohio). Although tests for HIV infection were not performed at the time of diagnosis in any of the cases, none of the patients had risk factors for HIV infection. By the clinical and pathological findings, patients with other secondary causes of FGS, and underlying primary or secondary forms of glomerulonephritis that might cause segmental glomerulosclerosis, were excluded from this study. In addition, cases considered as collapsing glomerulopathy were excluded by characteristic clinical features of severe nephrotic syndrome and rapid renal dysfunction, and by histology showing prominent tuft collapse with marked glomerular epithelial hyperplasia accompanied by micro-cystic tubuli with interstitial mononuclear cell infiltration (Valeri et al, 1994; Weiss et al, 1986).
Various features of epithelial pathology are included in FGS, even within the same patient. To define homogenous pathological lesions, we classified biopsies into two categories: 1) monolayered, polarized epithelial cells covering tuft sclerosis (monolayer lesion), and 2) two or more layers of epithelial cells with vacuolation and cytoplasmic droplets covering the GBM and filling the urinary space. This latter epithelial lesion was initially defined as “cellular lesions” by (Schwartz and Lewis 1985). Data in each category for all of the patients examined were accumulated and analyzed separately.
Immunohistochemical Staining
Immunohistochemical staining was performed on serial sections by a streptavidin-biotin technique described previously (Nagata et al, 1995a). Immunostaining for Ki-67 (DAKO, Kyoto, Japan), a marker of cells in the proliferative phase, and for p27Kip1 (Transduction Laboratories, Lexington, Kentucky), p57Kip2 (Santa Cruz Biochemicals, Santa Cruz, California), and cyclin A (Santa Cruz), cell-cycle molecules, was carried out after microwave pretreatment of the sections at 95° C, for 10 minutes. The specificity of these antibodies was previously characterized by Western blot analysis (Gerdes et al, 1991; Lloyd et al, 1997; Nagata et al, 1998b). Incubation with the first antibody was performed at 4° C overnight. Immunoreaction products were developed by using 3,3′-diaminobenzidine in PBS with 1% H2O2 or 3-amino-9-ethyl carbazole (AEC, DAKO). Sections were counterstained with hematoxylin or periodic acid-Schiff reagent when appropriate. PHM-5, an antibody that recognizes sialoprotein in the apical membrane of human podocytes, was generously provided by Dr. R.C. Atkins. The specificity of this antibody was previously described (Hancock and Atkins, 1983; Kerjaschki et al, 1986). PHM-5 was detected in detached podocytes that appeared in the urine of nephritis patients, indicating that PHM-5 is a useful marker even in damaged podocytes (Hara et al, 1995). Antibody against synaptopodin (PROGEN, Heidelberg, Germany), an actin-related protein specific for the podocyte foot process, and its specificity were previously reported (Mundel et al, 1997). Despite being called “epithelial cells,” mature differentiated podocytes do not express cytokeratin. The anti-cytokeratin antibody used in this study (C-2562; Sigma, St. Louis, Missouri) (Lane and Alexander, 1990) was exclusively negative in podocytes, but frequently positive in PEC in normal glomeruli and cellular crescents, which are known as a feature of PEC proliferation (Nitta et al 1999). Thus we chose cytokeratin as a marker of PEC phenotype. CD 68 (DAKO), an antibody specific for monocytes/macrophages, was used to determine whether epithelial lesions included monocytes/macrophages as found in cellular crescents in glomerulonephritis (Pulford et al, 1989). Its specificity and consistent immunoreaction with human renal tissue were described previously (Nagata et al, 1995a).
As a negative control for immunostaining, a normal portion of the renal tissue from renal cancer nephrectomy material was fixed by the same procedures as applied to the biopsy material and treated with either normal mouse serum or PBS alone. The control tissue was completely negative for immunostaining (data not shown).
References
Bariety J, Nochy D, Mandet C, Jaquot C, Glotz D, and Meyrier A (1998). Podocytes undergo phenotypic changes and express macrophage associated markers in idiopathic collapsing glomerulopathy. Kidney Int 53:918–925.
Barisoni L, Kriz W, Mundel P, and D’Agati V (1999). The dysregulated podocyte phenotype: A novel concept in the pathogenesis of collapsing ideopatic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 10:51–61.
Cameron JS (1996). The enigma of focal segmental glomerulosclerosis. Kidney Int Suppl 57:S119–S131.
Churg J, Habbib R, and White RHR (1970). Pathology of the nephrotic syndrome in children: A report for the International Study of Kidney Disease in Children. Lancet 760:1299–1302.
Churg J and Sorbin LH (1982). Focal segmental glomerulosclerosis. In: Churg J, editor. Classification and atlas of glomerular diseases, 2nd ed. Tokyo:IGAKU-SHOIN, 42–43.
Combs HL, Shankland SJ, Setzer SV, Hudkins KL, and Alpers CE (1998). Expression of the cyclin kinase inhibitor, p27Kip1, in developing and mature human kidney. Kidney Int 53:892–896.
D’Agati V (1994). The many masks of focal segmental glomerulosclerosis. Kidney Int 46:1223–1241.
Dantal J, Bigot E, Bogers W, Testa A, Kriaa F, Jaques Y, Hurault de Ligny B, Niaudet P, Charpentier B, and Soulillou JP (1994). Effect of plasma protein adsorption on protein excretion in kidney-transplant recipients with recurrent nephrotic syndrome. N Engl J Med 330:7–14.
Gerdes J, Li L, Schlueter C, Duchrow M, Wohlenberg C, Gerlach C, Stahmer I, Kloth S, Brandt E, and Flad HD (1991). Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am J Pathol 138:867–873.
Grishman E and Churg J (1975). Focal glomerular sclerosis in nephrotic patients: An electron microscopic study of glomerular podocytes. Kidney Int 7:111–122.
Hancock WV and Atkins RC (1983). Monoclonal antibodies to human glomerular cells: A marker for glomerular epithelial cells. Nephron 33:83–90.
Hara M, Yamamoto T, Yanagihara T, Takada T, Itoh M, Adachi Y, Yoshizumi A, Kawasaki K, and Kihara I (1995). Urinary excretion of podocalyxin indicates glomerular epithelial cell injuries in glomerulonephritis. Nephron 69:397–403.
Hattori M, Horita S, Yoshioka T, Yamaguchi Y, Kawaguchi H, and Ito K (1997). Mesangial phenotypic changes associated with cellular lesions in primary focal segmental glomerulosclerosis. Am J Kidney Dis 30:632–638.
Hiromura K, Pippin JW, Fero ML, Roberts JM, and Shankland SJ (1999). Modulation of apoptosis by the cyclin-dependent kinase inhibitor p27Kip1. J Clin Invest 103:597–604.
Kerjaschki D, Poczewski H, Dekan G, Horvat R, Balzar E, Kraft N, and Atkins RC (1986). Identification of a major sialoprotein in the glycocalyx of human visceral epithelial cells. J Clin Invest 78:1142–1149.
Kihara I, Tsuchida S, Yaoita E, Yamamoto T, Masanori H, Yanagihara T, and Takada T (1997). Podocyte detachment and epithelial cell retraction in focal segmental glomerulosclerosis with cellular variant. Kidney Int Suppl 63:S171–S176.
Kim YG, Alpers CE, Brugarolas J, Johnson RJ, Couser WG, and Shankland SJ (1999). The cyclin kinase inhibitor p21CIP1/WAF1 limits glomerular epithelial cell proliferation in experimental glomerulonephritis. Kidney Int 55:2349–2361.
Kriz W, Elger M, Nagata M, Kretzler M, Uiker S, Koeppen-Hageman I, Tenchert S, and Lemley K (1994). The role of podocytes in the development of glomerular sclerosis. Kidney Int Suppl 45:S64–S72.
Kriz W, Haenel B, Rosener S, and Elger M (1995). Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int 48:1435–1450.
Lane EB and Alexander CM (1990). Use of keratin antibodies in tumor diagnosis. Semin Cancer Biol 1:165–179.
Lloyd RV, Jin L, Quian X, and Kulig E (1997). Aberrant p27Kip1 expression in endocrine and other tumors. Am J Pathol 150:401–407.
Moll R, Hage C, and Thoenes W (1991). Expression of intermediate filament proteins in fetal and adult human kidney: Modulation of intermediate filament patterns during development and in damaged tissue. Lab Invest 65:74–86.
Mundel P, Heid HW, Mundel T, Krueger M, Reiser J, and Kriz W (1997). Synaptopodin: An actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol 139:193–204.
Nagata M, Akioka Y, Tsunoda Y, Komatsu Y, Kawaguchi H, Yamaguchi Y, and Ito K (1995a). Macrophages in childhood IgA nephropathy. Kidney Int 48:527–535.
Nagata M, Hattori M, Hamano Y, Ito K, Saitou K, and Watanabe T (1998a). Origin and phenotypic features of hyperplastic epithelial cells in collapsing glomerulopathy. Am J Kidney Dis 32:962–969.
Nagata M, Nakayama K, Terada Y, Hoshi S, and Watanabe T (1998b). Cell cycle regulation and differentiation of the human podocyte lineage. Am J Pathol 153:1511–1520.
Nagata M, Yamaguchi Y, and Ito K (1993). Loss of mitotic activity and the expression of vimentin in glomerular epithelial cells of developing human kidneys. Anat Embryol (Berl) 187:275–279.
Nagata M, Yamaguchi Y, Komatsu Y, and Ito K (1995b). Mitosis and presence of binucleate cells among glomerular podocytes in diseased human kidneys. Nephron 70:68–71.
Nitta K, Horita S, Honda K, Uchida K, Watanabe T, Nihei H, and Nagata M (1999). Glomerular expression of cell-cycle-regulatory proteins in human crescentic glomerulonephritis. Virchows Archiv 435:422–427.
Pabst R and Sterzel RB (1983). Cell renewal of glomerular cell types in normal rats. An autoradiographic analysis. Kidney Int 24:626–631.
Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, and Massague J (1994). Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78:59–66.
Pulford KA, Rigney EM, Micklem KJ, Jones M, Stross W, Gatter KC, and Mason DY (1989). KP1: A new monoclonal antibody that detects a monocyte/macrophage associated antigen in routinely processed tissue sections. J Clin Pathol 42:414–421.
Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis E, Lovell H, Warady B, Gunwar S, Chonko AM, Artero M, Vincenti F, Fine R, Wood E, Trachtman H (1996). Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med 334:878–883.
Schwartz MM and Lewis EJ (1985). Focal segmental glomerular sclerosis: The cellular lesion. Kidney Int 28:968–974.
Schwartz MM, Evans J, Bain R, and Korbet SM (1999). Focal segmental glomerulosclerosis: Prognostic implications of the cellular lesion. J Am Soc Nephrol 10:1900–1907.
Shankland ST (1997a). Cell-cycle control and renal disease (Review). Kidney Int 52:294–308.
Shankland ST, Floege J, Thomas SE, Nangaku M, Hugo C, Pippin J, Hennke K, Hockenberry DM, Johnson RJ, and Couser WG (1997b). Cyclin kinase inhibitors are increased during experimental membranous nephropathy: Potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int 52:404–413.
Sharif K, Goyal M, Kershaw D, Kunkel R, and Wiggins R (1998). Podocyte phenotypes as defined by expression and distribution of GLEPP1 in the developing glomerulus and in nephrotic glomeruli from MCD, CNF, and FSGS. Exp Nephrol 6:234–244.
Shimizu A, Masuda Y, Kitamura H, Ishizaki M, Sugisaki Y, and Yamanaka N (1996). Apoptosis in progressive crescentic glomerulonephritis. Lab Invest 74:941–951.
Valeri AV, Barisoni A, Appel GB, Seigle R, and D’Agati V (1994). Idiopathic collapsing focal segmental glomerulosclerosis: A clinicopathologic study. Kidney Int 50:1734–1746.
Weiss MA, Daquioag E, Margolin G, and Pollak V (1986). Nephrotic syndrome, progressive irreversible renal failure, and glomerular “collapse”: A new clinicopathological entity? Am J Kid Dis 7:20–28.
Acknowledgements
The authors wish to thank Dr. R. C. Atkins (Melbourne) for the generous gifts of antibody. The expert technical support for electron microscopy given by M. Kawashima is gratefully acknowledged. We also thank Professor Julie Vernon-Edo for the critical reading and English editing of the manuscript.
This work was supported by grants from the Ministry of Education, Science, and Culture of Japan (No. 10670154) and from the University of Tsukuba Research Projects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nagata, M., Horita, S., Shu, Y. et al. Phenotypic Characteristics and Cyclin-Dependent Kinase Inhibitors Repression in Hyperplastic Epithelial Pathology in Idiopathic Focal Segmental Glomerulosclerosis. Lab Invest 80, 869–880 (2000). https://doi.org/10.1038/labinvest.3780091
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780091
This article is cited by
-
Focal segmental glomerulosclerosis; why does it occur segmentally?
Pflügers Archiv - European Journal of Physiology (2017)
-
The emergence of the glomerular parietal epithelial cell
Nature Reviews Nephrology (2014)
-
Aberrant Notch1-dependent effects on glomerular parietal epithelial cells promotes collapsing focal segmental glomerulosclerosis with progressive podocyte loss
Kidney International (2013)
-
An in-depth analysis of proteomics expression profiling in rat glomeruli utilizing LC-MS
Chinese Science Bulletin (2010)
-
The enigmatic parietal epithelial cell is finally getting noticed: a review
Kidney International (2009)
