
Abstract
Malignant peripheral nerve sheath tumors (MPNST) are the most aggressive cancers associated with neurofibromatosis type 1 (NF1). Here we report a practical and reproducible model of intraneural NF1 MPNST, by orthotopic xenograft of an immortal human NF1 tumor-derived Schwann cell line into the sciatic nerves of female scid mice. Intraneural injection of the cell line sNF96.2 consistently produced MPNST-like tumors that were highly cellular and showed extensive intraneural growth. These xenografts had a high proliferative index, were angiogenic, had significant mast cell infiltration and rapidly dominated the host nerve. The histopathology of engrafted intraneural tumors was consistent with that of human NF1 MPNST. Xenograft tumors were readily examined by magnetic resonance imaging, which also was used to assess tumor vascularity. In addition, the intraneural proliferation of sNF96.2 cell tumors was decreased in ovariectomized mice, while replacement of estrogen or progesterone restored tumor cell proliferation. This suggests a potential role for steroid hormones in supporting tumor cell growth of this MPNST cell line in vivo. The controlled orthotopic implantation of sNF96.2 cells provides for the precise initiation of intraneural MPNST-like tumors in a model system suitable for therapeutic interventions, including inhibitors of angiogenesis and further study of steroid hormone effects on tumor cell growth.
Similar content being viewed by others

Main
Malignant peripheral nerve sheath tumors (MPNST) are often associated with neurofibromatosis type 1 (NF1), and are thought to arise from plexiform neurofibromas.1, 2 In fact, neurofibromas coexisting with MPNSTs were found in 81% of patients with NF1 but only in 41% of non-NF1 patients.3 Progression to malignancy from plexiform neurofibroma occurs in about 6% of NF1 patients, although the lifetime risk of MPNST in NF1 has been estimated as high as 8–13%,4 and is associated with high mortality.5
NF1 MPNSTs have distinctive characteristics.6 They are densely hypercellular and composed of spindle-shaped cells. The clonal elements with Schwann cell (SC) characteristics have a high proliferative index (5–38% Ki67-positive cells) and exhibit nuclear hyperchromasia and nuclear enlargement. Their growth is characterized by abrupt variation in cellularity and tissue pattern. They are firm, gray-tan and opaque, may grow very large and can be surrounded by a pseudocapsule. They may have areas of localized necrosis and tend to extend intraneurally.
A great deal of progress has been made developing cell lines and mouse models of NF1 tumors for experimental study and clinical testing. These cell lines and models have proven invaluable in furthering our understanding of the biology of NF1. Mice generated with a null mutation in the Nf1 gene die in utero by day 13. Targeted mutations in one Nf1 allele produce heterozygous mice (Nf1+/−) that genetically resemble the constitutional background of human NF1 patients, but fail to develop neurofibromas.7, 8 Chimeric mice carrying Nf1−/− cell populations9 and Nf1+/−; p53+/− crossed mice10 develop clonal neoplasms in peripheral nerves and have proven useful in studying genetic contributions to tumorigenesis. Conditional Nf1 knockout mice have been produced that develop neurofibromas when the knockout is in SCs.11 This model has significantly advanced the contention that SCs are the tumorigenic cells in NF1 neurofibromas. Xenografts of human tumors cell lines have been a mainstay of cancer research, as a way to recapitulate human tumor cell growth in vivo. Although not perfect models, xenografts have been and continue to be used extensively to study human tumor cell growth and to test potential therapeutics. Although a number of useful xenograft models have been used to test therapeutic treatments,12, 13, 14 none has shown true intraneural tumor growth closely resembling human NF1 MPNSTs. However, xenografts of human NF1 tumor cells have been shown to persist when injected into scid mouse sciatic nerves,15 and have recently been shown to recapitulate NF1 plexiform neurofibromas.16
An association between steroid hormones and neurofibromas has been previously hypothesized, based on reports of increased numbers and size of neurofibromas during puberty and pregnancy.17, 18 Dermal neurofibromas usually begin appearing in puberty, with general progression in numbers and size throughout much of adult life. Both plexiform and dermal neurofibromas can become larger during pregnancy, although some of these recede after pregnancy.19 This suggests that steroid hormones, which are elevated during puberty and pregnancy, may play a role in this ‘aggravation’ of neurofibroma growth. Since MPNSTs arise from existing plexiform neurofibromas in NF1, it is conceivable that the growth of some MPNSTs may also be influenced by steroid hormones, as are a number of other tumor types. Finally, to facilitate assessment of potential therapeutic testing in mouse models, in vivo imaging of experimental tumor growth and angiogenesis is of great interest.20, 21, 22, 23
Here we report that orthotopic xenografts of a human NF1 tumor-derived SC line into female scid sciatic nerves form large, intraneural, MPNST-like tumors rapidly, consistently and reproducibly in a site-specific manner. The growth and angiogenesis of these intraneural tumors can be monitored in vivo by magnetic resonance imaging (MRI) several weeks after initiation. We then apply this model to characterize the effects of steroid hormones on the in vivo intraneural growth of this cell line. The use of a permanent, commercially available cell line and standard methodology provide for high reproducibility by different laboratories and a valid working model for comparable study and testing of therapeutic approaches for NF1 MPNSTs.
MATERIALS AND METHODS
Originative Specimen and NF1 Cell Line
The NF1 tumor cell line, sNF96.2, was derived from tumor tissue resected from the leg of a 27-year-old male patient, who met the NF1 diagnostic criteria.24 This NF1 patient had a positive family history of NF1 and presented with multiple dermal neurofibromas, two spatially distinct plexiform neurofibromas, multiple café-au-lait spots, mild scoliosis and developmental delay. The portion of the tumor specimen used for tissue culture was independently characterized by immunohistopathology as an MPNST. The tissue was acquired with patient consent and used according to IRB-approved protocols.
DNA was extracted from blood leukocytes and tumor tissue as previously described by Colman et al.25 The sNF96.2 tumor cell line was established by methods described previously.15, 26 Briefly, tumor pieces were minced and dissociated for 3–5 h with dispase (1.25 U/ml; Collaborative Research) and collagenase (300 U/ml; type XI, Sigma) in L15 medium containing 10% calf serum and antibiotics. The digested tissue was dispersed by trituration and strained through a 30-mesh nylon screen. Collected cells were seeded on laminin-coated dishes and grown in DMEM containing 10% fetal bovine serum (FBS), 5% calf serum, glial growth factor-2 (25 ng/ml) and antibiotics. Cultures were subsequently grown and rapidly expanded without laminin and glial growth factor-2. Initial culture showed a homogenous SC-like population and a clonal morphology, which was retained through protracted passages. The apparently immortal cell line has spindle-shaped morphology and is immunopositive for S-100 and faintly positive for p75 (low-affinity neurotrophin receptor), indicating SC lineage. The cell line was deposited in the American Type Culture Collection.
NF1 Mutation Analysis
NF1 exons from tumor DNAs were analyzed by heteroduplex and SSCP analysis,27 as well as by direct sequencing.27 Samples were analyzed for loss of heterozygosity (LOH) using standard methods for genotyping NF1 polymorphisms as described previously by Colman et al25 and Rasmussen et al.28 Blood and tumor DNA results were compared when constitutional heterozygosity was seen at a given marker. In addition, standard cytogenetic analysis was performed on the tumor-derived SC cultures.
Mouse Strains
Immunodeficient B6 scid mice were used as hosts to minimize immunologic rejection of the xenografted human cell line. The scid, nonsense mutation in the DNA-PKCS gene, was described in by Blunt et al.29 Based on genomic DNA sequence (GenBank AB005213) PCR primers were designed flanking the mutation site in exon 85: scid 5, GAGTTTTGAGCAGACAATGCTGA and scid 3, CTTTTGAACACACACTGATTCTGC. The resulting 180-bp PCR product was digested with AluI to distinguish wild-type allele from mutant allele extra cut site via agarose gel electrophoresis, to genotype animals at the scid locus.
Tumor Xenografting
Xenografts in these studies were made by injecting human NF1 tumor-derived sNF96.2 cells into the sciatic nerves of adult female scid mice. sNF96.2 cultures from cryopreserved stocks were grown in DMEM containing 10% FBS and antibiotics. Dissociated cells were collected, rinsed thoroughly and resuspended at 1 × 108 cells/ml in calcium- and magnesium-free Hank's buffered salt solution. Young adult mouse hosts were anesthetized with isoflurane and the sciatic nerves exposed bilaterally at mid-thigh. A cell suspension (5 × 105 cells in 5 μl) was injected gradually into the sciatic nerve through a FlexiFil (0.2 mm OD) titanium-needle syringe (World Precision Instruments, Sarasota, FL, USA) driven by a UMII microinjector mounted on a motorized micromanipulator (World Precision Instruments). These techniques are for optimized tumor cell injection, but successful xenografts can be accomplished by hand and with simple equipment, as they were in our initial studies. The surgical site was then closed in layers with sutures and the revived mouse returned to specific pathogen-free housing. At 2–8 weeks after implantation, the animals were terminated under anesthesia and the nerves were removed and fixed by immersion in 4% paraformaldehyde. Xenograft success rate in female mice, based on the appearance of human glutathione S-transferase (huGST)-immunopositive tumors after 8 weeks, was approximately 93% (13 out of 14 xenografts, including initial studies).
To study the in vivo effects of steroid hormones on sNF96.2 tumor cell growth, a subset of ovariectomized female mice were given steroid hormones before xenografting, as described. Under anesthesia, the ovaries of female mice were removed. Next, these mice (n=3) were implanted subcutaneously with 60-day release pellets containing either 17 α-estradiol (0.72 mg/pellet), progesterone (25 mg/pellet) or placebo (Innovative Research of America, Sarasota, FL, USA), as per the manufacturer's instructions. Xenografts of sNF96.2 cells were carried out bilaterally, as described above. After 8 weeks, nerves were removed, embedded in paraffin and sectioned for staining. All animal use was performed in accordance to the guidelines of the University of Florida Animal Care and Use Committee.
Western Blot Analysis
Western Blots were performed as described by Muir et al.15 In these studies, neurofibromin was detected using a number of antibodies. We used the antibody available from Santa Cruz Biotechnology, raised against a peptide corresponding to residues 509–528 of the predicted NF1 gene product. To further investigate the possible effects of truncated NF1 gene products, we have developed monoclonal antibodies (McNFn27a, McNFn27b) raised against a peptide corresponding to the N-terminal residues 27–41 of the predicted NF1 gene product. Similar results were obtained with all antibodies.
Immunohistochemistry
Cell cultures
sNF96.2 monolayer cultures were examined for immunoreactivity to the SC antigens S-100 and the low-affinity neurotrophin receptor (p75), as described by Muir et al,15 with the following modifications: bound primary antibodies were labeled with swine anti-rabbit Igs (DAKO) (1/200) conjugated with fluorescein for 1 h at 37°C diluted in blocking buffer in darkness, and post-fixed with 2% paraformaldehyde in PBS for 10 min. After washing with PBS, slides were coverslipped and kept in the dark at 4°C until imaging. Imaging was performed using an excitation wavelength of 450–490 nm and an emission wavelength of 515–565 nm.
Xenografts
Processing and immunohistochemistry of sNF96.2 xenografts was carried out as described by Perrin et al.16
Magnetic Resonance Imaging
In vivo H-1 MRI was performed on mouse sciatic nerves, at the time points indicated, on a 4.7-T, 33-cm bore Avance magnet system (Bruker Instruments Inc., Billerica, MA, USA) imaging spectrometer at 200 MHz. Mice were anesthetized with isoflurane, positioned in the RF coil and placed in the MRI system. Fat-suppressed T1-weighted (repetition time (TR) 1000 ms, echo time (TE) 10.5 ms, number of averages (NA) 4) and fat-suppressed T2-weighted (TR 3000 ms, TE 60 ms, NA 6) spin-echo images were collected with a field-of-view (FOV) of 40 × 20 mm in a matrix of 192 × 96 as 0.5-mm slices transverse to the spine. The legs were positioned so that the femurs lie in the image plane (see Figure 6). Increased vascular permeability, indicating angiogenesis was visualized by dynamic contrast-enhanced (DCE)-MRI. For DCE-MRI, fat-suppressed T1-weighted images (same parameters as above, except TR 400 ms, TE 7.3 ms, NA 8. matrix 128 × 64) were obtained using a high-molecular-weight contrast agent gadolinium diethylenetriaminepenta-acetate (Gd-DTPA). At the time points indicated, three DCE MR images were collected before intraperitoneal injection of 0.3 mmol/kg Gd-DTPA, and 10 images were collected after injection. The contrast agent was seen first as a hyperintensity in tissues with the highest level of vascular permeability. The experiment shown was performed on a mouse that was only xenografted on one sciatic nerve, the other being left untouched, and was representative of other similar experiments. Regions of interest were chosen to represent the xenografted tumor, normal nerve and muscle. All MR images were processed using software routines written in IDL (Research Systems Inc., Boulder, CO, USA).
RESULTS
Genetic and Phenotypic Characterization of the sNF96.2 Cell Line
Sample sNF96.2 had an abnormal karyotype, which is typical for MPNSTs.30 There were clonal findings of 48, X, −X or Y, +7, add(7)(p22)x2, +8, add(9)(p24), +mar[10] in 10/10 metaphase cells. There were also a few non-clonal rearrangements; however, both chromosome 17 s looked normal. The NF1 germline mutation was identified as a base pair deletion in exon 21 (3683delC), causing a frameshift, which leads to a premature stop codon before the ras-GAP domain. In addition, somatically, this tumor had LOH of the entire 17 homologs31 and the genotyping showed complete LOH (no detection of the remaining allele). Thus, this culture would not be expected to produce any full-length neurofibromin protein. Further, the SC cultures derived from the sNF96.2 sample had LOH for a marker in the p53 gene with the entire chromosome 17 homologs missing and the one carrying the NF1 mutation reduplicated (data not shown). Immunostaining of the primary sNF96.2 tumor showed weak but unremarkable p53 staining (data not shown). sNF96.2-cultured cells also showed no abnormalities on SSCP analysis of exons 5–8, which are most often involved in tumorigenesis.
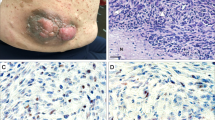
As predicted by genotyping, the SC cultures derived from the sNF96.2 sample showed no full-length neurofibromin (Mr≈250 kDa) when extracts were analyzed by Western immunoblotting using several anti-neurofibromin antibodies (Figure 1). Similarly, full-length neurofibromin was not detected in the extracts of fibroblast cultures derived from embryonic Nf1−/− knockout mice. As a positive control, normal human SC cultures showed a substantial band-pair corresponding to full-length neurofibromin. A secondary immunolabeling of the blot for huGST showed consistent loading for all human samples. In monolayer culture, a majority of sNF96.2 cells were spindle-shaped and faintly immunopositive for the SC marker S-100 (Figure 2a) and the low-affinity nerve growth factor receptor p75 (Figure 2c). sNF96.2 xenograft tumors were distinctly S-100- and p75-immunopositive, as were the normal portion of the host mouse nerve, indicating the SC-like phenotype was retained in vivo (Figure 2b and d, respectively). Finally, it has previously been shown that the primary sNF96.2 tumor sample has increased estrogen and progesterone receptor, as compared with normal human SCs. However, no increase was observed on the derived sNF96.2 cell line.32

sNF96.2 cells fail to express full-length neurofibromin. Extracts from sNF96.2 cultures, fibroblast cultures derived from embryonic Nf1−/− knockout mice, and normal human SC cultures were analyzed for neurofibromin expression by Western immunoblotting. Full-length neurofibromin appeared as a ≈250-kDa band in the normal human SC sample, but was absent in the sNF96.2 and Nf1−/− knockout fibroblasts. An equal amount of total protein was loaded for each sample. Immunblotting for huGST was performed to demonstrate consistent protein loading.

sNF96.2 cells have a Schwann-like phenotype. sNF96.2 cultures (a, c) were examined for the immunoexpression of two SC markers, S-100 (a) and p75 (c) by fluorescent immunocytochemistry. p75 was easily detected on the surface of sNF96.2 cells, while only faint cytoplasmic labeling for S-100 was observed. Distinct immunoexpression of these SC markers by sNF96.2 xenograft tumors was observed by immunoperoxidase methods (n, normal nerve; t, xenograft tumor) (Figure 2b and d, respectively). Original magnifications: (a, c): × 400; (b, d): × 200.
sNF96.2 Xenografts Form Massive MPNST-Like Tumors
We previously found that a subset of neurofibromin-negative NF1 tumor cultures form slow-growing tumors as intraneural xenografts. In addition, normal human SC intraneural xenografts showed only transient occupancy, limited survival and were often undetectable after 8 weeks in vivo.15 Using the same methods, intraneural xenografts of the sNF96.2 SC line grew extraordinarily well and rapidly progressed to MPNST-like tumors in over 88% of female mice tested over various time points (n=17). The resulting masses were firm, gray-tan in color, grew rapidly and were histopathologically similar to human NF1 MPNSTs. Xenografts also grew in male scid mice, but did not progress as dramatically or as quickly as they did in female hosts. The results presented in this report exclusively represent orthotopic sNF96.2 xenografts in adult female scid mice after 8 weeks (n=5). Figure 3a shows the gross morphology of a normal mouse sciatic nerve and a representative large sNF96.2 xenograft 8 weeks after engraftment. Orthotopic xenografts of sNF96.2 cells resulted in an average increase in nerve diameter of over three-fold (0.99 mm±0.33, n=5) when compared with normal, age-matched mouse sciatic nerves (0.304 mm±0.0392, n=5). Control injections of vehicle alone indicated the increase in nerve diameter did not result from the needle injection per se. Vehicle injected mouse sciatic nerves were not increased in size compared to normal nerves (0.293 mm±0.0645, n=2 vs 0.304 mm±0.0392, n=5, respectively). As a further control, normal human SCs were injected using the same procedures. These control xenografts resulted in only a slight, 9.7% increase in nerve diameter (0.334 mm±0.0643, n=6 vs 0.304 mm±0.0392, n=5) after 8 weeks. Therefore, the increase in nerve diameter observed consistently in sNF96.2 xenografts resulted from the characteristic intraneural growth of these cells, and was not caused by the needle injection or injury response.

sNF96.2 xenograft tumors. Eight weeks after implantation, sNF96.2 tumor xenografts grossly enlarge in the sciatic nerve of a female scid host (lower). A normal mouse sciatic nerve (upper) is shown as a reference. Scale bar=2 mm.
These mice developed large, rapidly growing, bulky tumors that caused intraneural disruption and host nerve remodeling. Histological staining (Figure 4a) showed these tumors consistently displayed a dense hypercellularity and were composed of elongated spindle-shaped cells, typical of human MPNST. Other characteristics of human MPNST observed in xenograft tumors include nuclear hyperchromasia, mitoses and a fascicular growth pattern. To prove the tumors resulted from the expansion of the xenografted human tumor cells, engrafted nerves were immunostained with an antibody that labels exclusively huGST (Figure 4b and e). HuGST-positive cells were found throughout and were by far the major component of the tumor masses. Immunopositive sNF96.2 cells increased in number over time and expanded intraneurally, often dominating the nerve completely. In contrast to sNF96.2 SCs, normal human SC xenografts showed only transient occupancy, severely limited survival and were undetectable in four out of six (67%) xenografts after 8 weeks.15 In the two mice that did display positive huGST staining, survival of xenografted normal human SCs was severely limited and there was only meagre, transient occupancy.15 Immunostaining with an antibody specific to human Ki67 (a nuclear antigen found in proliferating cells), but that does not cross react with mouse tissue (data not shown), indicated after 8 weeks, sNF96.2 xenografts had a very high rate of proliferation. Almost half of the cells stained positive for Ki67 (42.25±4.85% Ki67-positive cells per high-power field, n=5) (Figure 4c). Ki67-positive nuclei were seen only in the areas of the nerve occupied by human xenografted cells and not in adjacent host nerve tissue, confirming the specificity of this analysis. These results indicate that the female scid mouse nerve provides a favorable environment for the development of MPNST-like tumors by xenografts of the sNF96.2 SC line. The vascularity of sNF96.2 tumors was examined by immunostaining for von Willebrand's factor (vWF), a marker of mature endothelial cells. Immunostaining for vWF revealed scant blood vessels in normal mouse nerves arranged almost exclusively along the longitudinal nerve axis (data not shown). Xenografted tumors were highly vascularized compared with normal mouse sciatic nerves (Figure 4d). Nearly identical results were obtained by labeling xenograft tumor tissue with antibodies to Flk-1, a high-affinity receptor for vascular endothelial growth factor (VEGF) also found on blood vessels (data not shown). Aberrant vessel development was observed as early as 2 weeks after orthoptopic xenograft, and was closely associated with tumor hypercellularity. The vascular pattern in the tumors was irregular compared with the longitudinal alignment found in tumor-free areas of the host nerve and normal nerve, indicating extensive tissue remodeling. These results show the induction of new blood vessel formation in response to the growth of xenograft tumors and provide the opportunity to examine angiogenesis in this model of NF1 MPNST.

sNF96.2 xenografts are highly proliferative, angiogenic and infiltrated by mast cells. (a) Hematoxylin and eosin staining shows the xenograft tumors are dense, hypercellular masses. (b) HuGST immunostaining indicates the tumors have thoroughly occupied and displace the host nerve elements. (c) Ki67 immunostaining shows a very high proliferative rate. (d) Immunostaining for vWF labels the increased vascularity associated with the xenografts. (e) Xenografted nerves were immunostained for huGST (brown) and counterstained for mast cells using acidic toluidine blue. Compared with normal nerve, there was an overall increase in mast cell infiltration in the xenografted nerves. A few mast cells were found within the tumor mass (inset), however, the major accumulation occurred in the host nerve tissue surrounding the tumor (arrows). Original magnification, × 200.
Sections of sNF96.2 xenografts immunostained for huGST were counterstained with acidic toluidine blue to visualize mast cells and their spatial relationship to tumor development. While occasional mast cells were seen in normal scid mouse sciatic nerves, there was an overall increase in mast cell number in the xenografted nerves. A few of these infiltrating mast cells colocalized within the main tumor mass (ie, with intense huGST staining) (Figure 4e, inset). However, there were more mast cells in the tissues surrounding the huGST-positive areas (Figure 4e). The boundary with the non-staining mouse tissue and huGST-staining tumor is also evident (Figure 4e).
The data presented above represent xenografts developed in female mice. Interestingly, rapid and massive tumor growth by the sNF96.2 cell line occurred only in female hosts, while sNF96.2 tumors in male hosts grew much less vigorously. In fact, when examined after 4 weeks, only 33% (n=6) of the sNF96.2 xenografts in male hosts resulted in a discernible huGST-positive mass compared with over 88% (n=17) in female mice. Given time, tumors did sometimes develop in male nerves, but never to the degree or size that they did in female nerves. These results suggest a potential hormonal influence on the in vivo growth of this cell line. To determine whether female steroid hormones affect tumor cell growth in vivo, female mice were ovariectomized to attenuate the level of female steroid hormones. Blank pellets (placebo) or pellets containing estrogen or progesterone were then implanted subcutaneously to provide physiological levels of the respective hormones. Xenografts of sNF96.2 cells were carried out as described above and tumors were allowed to develop for 8 weeks. Assessment of tumor cell proliferation via Ki67 immunhistochemistry revealed that tumor cell proliferation dropped significantly in ovariectomized mice, compared with intact female mice. Replacement of estrogen or progesterone caused a significant increase in tumor cell proliferation, almost to levels measured in intact females (Figure 5). These results suggest a potentially supportive role for steroid hormones is NF1 tumor development.

Steroid hormones affect xenografted sNF96.2 tumor cell proliferation. Proliferation of xenografted sNF96.2 tumor cells was assessed by counting Ki67-positive cells in × 400 high-power fields (HPF) of tumor sections from normal mice, ovariectomized mice and ovariectomized mice receiving steroid hormone replacement. Reduction of steroid hormones via ovariectomy with no steroid hormone replacement (placebo) greatly reduced xenografted sNF96.2 tumor cell proliferation when compared with xenografts in mice with intact ovaries (*P<0.0001). Replacement of physiological levels of estrogen (**P=0.004) or progesterone (***P=0.003) increased xenografted sNF96.2 tumor cell proliferation in ovariectomized mice to almost normal levels. Statistical significance was calculated using the Student's t-test.
In Vivo MRI of Tumor Xenografts
An important feature of this NF1 xenograft model is the ability to precisely initiate tumors within the mouse sciatic nerve. This feature simplifies later detection and monitoring of tumor growth. To that end, we tested various MRI methods to examine tumor development in vivo. Normal mouse nerve showed little contrast in T1-weighted MRI, both in our experience and in published results.33, 34 On the other hand, xenografted tumors appeared as highly contrasted, hyperintense regions on in vivo T2-weighted MRI (Figure 6a). A slight hyperintensity was discernible at the site of tumor cell implantation 2 weeks after xenograft of sNF96.2 cells. By 4 weeks, a tumor mass was easily distinguished in nearly all xenografted nerves and tumor expansion was readily monitored thereafter. The hyperintense tumor images were subsequently verified as xenografted sNF96.2 cells by post-mortem huGST immunostaining (see Figure 4). Thus, T2-weighted, in vivo MRI is a powerful tool for monitoring tumor growth over time and can subsequently be used to test the effectiveness of therapeutic agents in vivo. To demonstrate increased vascular permeability, an assessment of angiogenesis, DCE-MRI was also performed 8 weeks after xenograft of sNF96.2 cells. DCE-MRI, where the uptake and washout of a contrast agent in tissues is monitored over time,35, 36 distinguished a highly contrasted, hyperintense region in the xenografted area of the nerve (Figure 6b). This region corresponded to the xenograft tumor mass shown in Figure 6a. Approximately 20 min after systemic injection of the contrast agent, when the level of contrast enhancement peaked (Figure 6c), the xenografted tumor displayed an average contrast enhancement eight-fold higher than the surrounding muscle, while the contralateral, normal nerve showed only an average two-fold increase over the surrounding muscle. This indicates some leakiness to gadolinium-contrast-agent across the blood–nerve barrier in the mouse normal nerve, but much more leakage occurred in the hypervascular xenografted tumors. Similar results were obtained in replicate mice xenografted with sNF96.2 cells. These results suggest an increased vascular permeability in the xenografted tumor, which correlates with the histological findings of tumor-induced angiogenesis.

sNF96.2 tumor growth and angiogenesis monitored by in vivo MRI. Xenografted mouse sciatic nerves were imaged in vivo on a 4.7T magnet at various times after tumor cell injection. (a) T2-weighted MRI reveals a large tumor mass (arrow) 8 weeks after orthotopic implantation of sNF96.2 cells. (b) T1-weighted DCE-MRI from the same mouse shown in (a) after systemic injection of the contrast agent gadolinium. The left image shows the contralateral sciatic nerve, which was not injected with tumor cells, located 2.5 mm proximal to the image on the right. DCE-MRI shows increased tumor blood vessel permeability. (c) The graph shows the percent enhancement in the specified regions of interest (indicated by the circles; n, normal nerve; t, xenograft tumor; m, muscle) over time after injection of the contrast agent (indicated by arrow). The percent enhancement of the tumor increased dramatically, while the regions in both the surrounding muscle and the contralateral, normal nerve rise only slightly. This increase in percent enhancement corresponds to increased vascular perfusion in xenografted tumor area associated with angiogenesis and increased tumor vascularity.
Interestingly, metastases were not observed in the xenografted host organs, and this was also true for the originative patient tumor. Although this might be somewhat unusual for an MPNST, the sNF96.2 xenografts were otherwise remarkably consistent with MPNST histopathology. Classifications for peripheral nerve sheath tumors arising in genetically engineered mouse (GEM) models have been devised because of some important differences between human and murine lesions.37 In the same way, it is difficult to apply the GEM classifications to tumors arising in xenografting models. Clearly, sNF96.2 xenograft tumors result from the intraneural proliferation of NF1-deficient SCs and the admixture of various cell types from the mouse nerve, including endothelial and mast cells. For the most part, sNF96.2 xenografts fit the Grade III PNST GEM tumor classification because of high cellularity, anaplasia and frequent mitotic figures.
DISCUSSION
It is estimated that about 6–13% of NF1 patients will develop MPNST and all patients with neurofibromas are at risk for malignant degeneration.38 Because surgical debulking is currently the main treatment option for NF1 tumors, there is a pressing need for consistent, clinically relevant models of NF1 MPNST. In fact, xenograft models have already been used to study therapeutic effects in intraperitoneal and subcutaneous model systems.12, 13, 14 Although useful for studying therapeutic effects on in vivo tumor growth, these models do not recapitulate the complex neural microenvironment found in human NF1 MPNSTs. Using a simple and reproducible xenografting technique, we have established a mouse model for intraneural NF1 MPNST applicable for practical therapeutic testing in a relevant cellular environment. When xenografted into the nerves of female immunodeficient mice, the NF1 MPNST cell line, sNF96.2, rapidly forms tumors bearing the characteristics of human NF1 MPNST. The sNF96.2 cell line is immortal, commercially available and apparently stable after protracted passage in vitro. Similar to Nf1-null cells cultured from embryonic knockout mice,39, 40 the sNF96.2 culture proliferates in a growth factor-independent manner. This cell line has a frameshift germline mutation, a complete LOH for NF1, LOH for a marker in the p53 gene and fails to express neurofibromin. Unlike SCs only deficient in neurofibromin, sNF96.2 cells are highly tumorigenic and have multiple genetic defects. These features are associated with highly autonomous growth properties. In that regard, a complex abnormal genotype is more characteristic of progressive NF1 tumors.
Intraneural sNF96.2 tumors exhibit histopathological characteristics of human NF1 MPNSTs. They are hypercellular with hyperchromatic nuclei, have a high proliferative index, increased numbers of blood vessels and show mast cell infiltration. The orthotopic nature of these xenografts produces a tumor representative of human NF1 MPNSTs. Uncharacteristic of many MPNSTs in NF1 patients, there were no obvious signs of necrosis or metastases in the 8-week time frame of these studies. Interestingly, this was also true for the originative tumor. Since these xenografts were only allowed to grow for 8 weeks, it is conceivable that tumors growing for longer times may develop areas of necrosis and metastases.
Angiogenesis is a common feature of many tumors and neurofibroma SCs have been previously shown to be angiogenic.41 Necrosis results from rapid tumor growth and a lack of adequate blood supply. Therefore, the sNF96.2 tumors, despite their rapid growth, appear to induce adequate angiogenesis that precludes necrosis. This provides an attractive therapeutic target, since inhibiting the growth of NF1 tumor blood vessels with antiangiogenic agents may slow, halt or even reverse tumor progression. Angiogenic factors have been shown to increase the permeability of tumor blood vessels (reviewed by Bates et al42), and thus allow monitoring of tumor angiogenesis with MRI using contrast agents.43 We established methods to monitor xenograft tumor growth and tumor-induced angiogenesis in vivo, using T2-weighted MRI and DCE-MRI, respectively. These techniques will be valuable in monitoring tumor progression in testing the effectiveness of potential therapeutic treatments. Further, MRI has been suggested as a potentially useful tool in detecting progression to malignancy in NF1 neurofibromas.44 We are currently exploring the use of MRI in studying the malignant progression of neurofibromas, using NF1 xenograft models of both MPNST and plexiform-like neurofibroma.
The scid mice used in these studies are immunocompromised (to prevent rejection of the implanted human tumor cells) yet do posses an intact innate immune system that includes mast cells.45 Mast cells have been implicated as important factors in tumor development and angiogenesis (reviewed by Folkman46 and Ribatti et al47), and are potent producers of matrix metalloproteinases and pro-angiogenic factors such as VEGF, basic fibroblast growth factor and transforming growth factor-β.48, 49 Also, the Nf1 status of mast cells may be important for tumor formation and progression in some Nf1 tumor models, and these studies have suggested that mast cells may induce or contribute to tumor formation in Nf1 mutant mouse models.11, 50 In addition, Nf1+/− mast cells were found to hyperproliferate in response to mast cell mitogens,51 and NF1 tumorigenic SCs produce a potent mast cell mitogen, stem cell factor.51 Previous studies have suggested that mast cells may induce or contribute to tumor formation in Nf1 mutant mouse models.11, 52 We found that mast cells infiltrate sNF96.2 xenografted tumors in scid mice, and do so in a manner similar to mast cell infiltrates seen in human NF1 MPNSTs.53 Mast cells may be important contributors to NF1 tumor angiogenesis and progression in the tumor environment.
An unexpected result of these studies was that, whereas tumors developed in both male and female mouse sciatic nerves, sNF96.2 tumors developed significantly better in female than in male hosts. These results suggest a possible in vivo hormonal influence in tumor cell growth of sNF96.2 cell xenografts. This is consistent with an anecdotal consensus that tumors in NF1 patients often become evident and more symptomatic during times of pronounced hormonal changes, such as puberty and pregnancy. Some patients have reported an increase in size and number of both dermal and plexiform neurofibromas during pregnancy18 or while taking birth control pills (MR Wallace, unpublished data). A recent survey of 59 NF1 patients found that oral contraceptive pills did not stimulate subjective growth of neurofibromas in the majority of patients, in contrast to two patients receiving high-dose depot contraceptive with progesterone, who did report significant tumor growth.54 There have even been reports of increased malignant potential of plexiform tumors during pregnancy.55, 56 This information suggests that steroid hormones may play a role in neurofibroma development, growth and/or survival. MPNST growth is not normally thought to be hormone-driven, but in this model, tumor cell proliferation appears to be supported by steroid hormones. Males are known to have circulating hormones, including estrogen and progesterone, which may have supported tumor growth in the original male patient. Unfortunately, no patient samples are available to test for the presence of these hormones. Also, sNF96.2 cells did grow and produce small tumors in male mice, although their size and rates of proliferation were comparatively much smaller than identical xenografts in female hosts (data not shown). While no detectable estrogen nor progesterone receptors were found on the sNF96.2 cell line, these receptors were found in the original patient tumor;32 and although no increase in sNF96.2 cell proliferation was observed in vitro in the presence of estrogen or progesterone,32 our results indicate that estrogen and progesterone each may support tumor cell proliferation in NF1 MPNST intraneural sNF96.2 xenografts. As suggested by Fishbein et al,32 tumor growth response to steroid hormones may be variable, probably being patient- or tumor-specific, and may be due to hormonal effects on cells surrounding the tumors. Given the differences in steroid hormone effects on sNF96.2 cells between the in vitro results shown by Fishbein et al32 and the in vivo results presented here, future studies may do well to examine the hormonal influences on cells of the tissue stroma rather than on the tumor cells themselves. Whether studying NF1 MPNSTs or another type of cancer, these results support the importance of the tumor microenvironment when considering animal models. We believe true orthotopic models of human tumors, such as the intraneural model presented here and elsewhere,16 have a great advantage in recapitulating the tumor growth characteristics of human tumors, especially for therapeutic testing. Clearly, the tumor environment in most types of cancers, whether cellular or systemic, can have a pronounced influence on tumor growth, including NF1 tumors.
The orthotopic xenograft model presented here is a practical, highly reproducible and representative model of intraneural NF1 MPNST, with a great potential for therapeutic testing. Estrogen and progesterone treatments were shown to reinstate the higher rates of proliferation in ovariectomized NF1 MPNST-like sNF96.2 xenografts, suggesting a possible supportive role for these steroid hormones in some NF1 MPNST tumorigenesis, suggesting this model could be useful for other steroid hormone studies. Interestingly, notable differences were observed when comparing our xenograft models of NF1 MPNST and NF1 plexiform-like neurofibroma.16 These differences were most evident in the rates of tumor development and interactions with the cellular and systemic environments. Current and future work will use these tumor models in scid mice also harboring Nf1 heterozygous mutations. These experiments will help ascertain the importance of the role of Nf1 heterozygosity in the tumor microenvironment, and it's role in tumorigenicity. Xenograft tumor models underscore the individualistic nature of NF1 tumors and reproduce the heterogeneity recognized in human NF1 neoplasia.
Accession codes
References
Friedman JM, Gutmann DH, MacCollum M, et al (eds). Neurofibromatosis: Phenotype, Natural History and Pathogenesis, 3rd edn. The Johns Hopkins Press: Baltimore, MD, 1999.
Woodruff JM . Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am J Med Genet 1999;89:23–30.
Ducatman BS, Scheithauer BW, Piepgras DG, et al. Malignant peripheral nerve sheath tumor. A clinicopathologic study of 120 cases. Cancer 1986;57:2006–2021.
Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39:311–314.
Korf BR . Neurofibromas and malignant tumors of the peripheral nerve sheath. In: Friedman JM, Gutmann DH, MacCollum M, Riccardi VM (eds). Neurofibromatosis: Phenotype, Natural History and Pathogenesis, 3rd edn. The Johns Hopkins Press: Baltimore, MD, 1999, pp 142–161.
Scheithauer BW, Woodruff JM, Erlandson RA . Tumors of the peripheral nervous system. In: Rosai J, Sobin LH (eds). Atlas of Tumor Pathology. Series 3, Fascicle 24. Armed Forces Institute of Pathology: Washington, DC, 1997, pp 385–405.
Brannan CI, Perkins AS, Vogel KS, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev Rev 1994;8:1019–1029.
Jacks T, Shih TS, Schmitt EM, et al. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 1994;7:353–361.
Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science 1999;286:2172–2176.
Vogel KS, Klesse LJ, Velasco-Miguel S, et al. Mouse tumor model for neurofibromatosis type 1. Science 1999;286:2176–2179.
Zhu Y, Ghosh P, Charnay P, et al. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 2002;296:920–922.
Mahller YY, Vaikunth SS, Currier MA, et al. Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol Ther 2007;15:279–286.
Hirokawa Y, Nakajima H, Hanemann CO, et al. Signal therapy of NF1-deficient tumor xenograft in mice by the anti-PAK1 drug FK228. Cancer Biol Ther 2005;4:379–381.
Hirokawa Y, Nheu T, Grimm K, et al. Sichuan pepper extracts block the PAK1/cyclin D1 pathway and the growth of NF1-deficient cancer xenograft in mice. Cancer Biol Ther 2006;5:305–309.
Muir D, Neubauer D, Lim IT, et al. Tumorigenic properties of neurofibromin-deficient neurofibroma Schwann cells. Am J Pathol 2001;158:501–513.
Perrin G, Fishbein L, Thomson S, et al. Plexiform-like neurofibromas develop in the mouse by intraneural xenograft of an NF1 tumor-derived Schwann cell line. J Neurosci Res 2007;85:1347–1357.
Riccardi VM . Type 1 neurofibromatosis and the pediatric patient. Curr Probl Pediatr 1992;22:66–106.
McLaughlin ME, Jacks T . Progesterone receptor expression in neurofibromas. Cancer Res 2003;63:752–755.
Dugoff L, Sujansky E . Neurofibromatosis type 1 and pregnancy. Am J Med Genet 1996;66:7–10.
Messerli SM, Tang Y, Giovannini M, et al. Detection of spontaneous schwannomas by MRI in a transgenic murine model of neurofibromatosis type 2. Neoplasia 2002;4:501–509.
Banerjee D, Hegedus B, Gutmann DH, et al. Detection and measurement of neurofibromatosis-1 mouse optic glioma in vivo. Neuroimage 2007;35:1434–1437.
Rosenbaum T, Engelbrecht V, Krolls W, et al. MRI abnormalities in neurofibromatosis type 1 (NF1): a study of men and mice. Brain Dev 1999;21:268–273.
Chang LS, Jacob A, Lorenz M, et al. Growth of benign and malignant schwannoma xenografts in severe combined immunodeficiency mice. Laryngoscope 2006;116:2018–2026.
Gutmann DH, Aynsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51–57.
Colman SD, Williams CA, Wallace MR . Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat Genet 1995;11:90–92.
Wallace MR, Rasmussen SA, Lim IT, et al. Culture of cytogenetically abnormal Schwann cells from benign and malignant NF1 tumors. Genes Chromosomes Cancer 2000;27:117–123.
Abernathy CR, Rasmussen SA, Stalker HJ, et al. NF1 mutation analysis using a combined heteroduplex/SSCP approach. Hum Mutat 1997;9:548–554.
Rasmussen SA, Colman SD, Ho VT, et al. Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 1998;35:468–471.
Blunt T, Gell D, Fox M, et al. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA 1996;93:10285–10290.
Mertens F, Dal Cin P, De Wever I, et al. Cytogenetic characterization of peripheral nerve sheath tumours: a report of the CHAMP study group. J Pathol 2000;190:31–38.
Rasmussen SA, Overman J, Thomson SA, et al. Chromosome 17 loss of heterozygosity studies in benign and malignant tumors in neurofibromatosis type 1. Genes Chromosomes Cancer 2000;28:425–431.
Fishbein L, Zhang X, Fisher LB, et al. In vitro studies of steroid hormones in neurofibromatosis 1 tumors and Schwann cells. Mol Carcinogen 2007;46:512–523.
Aagaard BD, Lazar DA, Lankerovich L, et al. High resolution magnetic resonance imaging is a noninvasive method or observing injury and recovery in the peripheral nervous system. Neurosurgery 2003;53:199–204.
Grant GA, Britz GW, Goodkin R, et al. The utility of magnetic resonance imaging in evaluating peripheral nerve disorders. Muscle Nerve 2002;25:314–331.
Tofts PS, Brix G, Buckley DL, et al. Estimating kinetic parameters from dynamic contrastenhanced T1-weighted MRI of a diffusible tracer: standardized quantities and symbols. J Magn Reson Imaging 1999;10:223–232.
Evelhoch JL . Key factors in the acquisition of contrast kinetic data for oncology. J Magn Reson Imaging 1999;10:254–259.
Stemmer-Rachamimov AO, Louis DN, Nielsen GP, et al. Comparative pathology of nerve sheath tumors in mouse models and humans. Cancer Res 2004;64:3718–3724.
Schwarz J, Belzberg AJ . Malignant peripheral nerve sheath tumors in the setting of segmental neurofibromatosis. Case report. J Neurosurg 2000;92:342–346.
Vogel KS, Brannan CI, Jenkins NA, et al. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell 1995;82:733–742.
Kim HA, Ling B, Ratner N . Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol 1997;17:862–872.
Sheela S, Riccardi VM, Ratner N . Angiogenic and invasive properties of neurofibroma Schwann cells. J Cell Biol 1990;111:645–653.
Bates DO, Hillman NJ, Williams B, et al. Regulation of microvascular permeability by vascular endothelial growth factors. J Anat 2002;200:581–597.
Medved M, Karczmar G, Yang C, et al. Semiquantitative analysis of dynamic contrast enhanced MRI in cancer patients: variability and changes in tumor tissue over time. J Magn Reson Imaging 2004;20:122–128.
Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis type 1: MRI supports the diagnosis of malignant peripheral nerve sheath tumor. Neuroradiology 2003;45:618–625.
Dorshkind K, Keller GM, Phillips RA, et al. Functional status of cells from lymphoid and myeloid tissues in mice with severe combined immunodeficiency disease. J Immunol 1984;132:1804–1808.
Folkman J . Role of angiogenesis in tumor growth and metastasis. Semin Oncol 2002;29 (6 Suppl 16):15–18.
Ribatti D, Crivellato E, Roccaro AM, et al. Mast cell contribution to angiogenesis related to tumour progression. Clin Exp Allergy 2004;34:1660–1664.
Qu Z, Liebler JM, Powers MR, et al. Mast cells are a major source of basic fibroblast growth factor in chronic inflammation and cutaneous hemangioma. Am J Pathol 1995;147:564–573.
Meininger CJ, Zetter BR . Mast cells and angiogenesis. Semin Cancer Biol 1992;3:73–79.
Yang F-C, Ingram DA, Chen S, et al. Neurofibromin-deficient Schwann cells secrete potent migratory stimulus for Nf1+/− mast cells. J Clin Invest 2003;112:1851–1861.
Ingram DA, Hiatt K, King AJ, et al. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med 2001;194:57–69.
Ryan JJ, Klein KA, Neuberger TJ, et al. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J Neurosci Res 1994;37:415–432.
Viskochil DH . It takes two to tango: mast cell and Schwann cell interactions in neurofibromas. J Clin Invest 2003;112:1791–1793.
Lammert M, Mautner V-F, Kluwe L . Do hormonal contraceptives stimulate growth of neurofibromas? A survey on 59 NF1 patients. BMC Cancer 2005;5:16–19.
Puls LE, Chandler PA . Malignant schwannoma in pregnancy. Acta Obstet Gynecol Scand 1991;10:243–244.
Posma E, Aalbers R, Kurniawan KS, et al. Neurofibromatosis type I and pregnancy: a fatal attraction? Development of malignant schwannoma during pregnancy in a patient with neurofibromatosis type I. BJOG 2003;110:530–532.
Acknowledgements
Supported by the National Institutes of Health Training Grant T32-CA09126-27 (GQP), and Grants R01 NS42075 and P41 RR16105 (THM), the US Department of Defense Grants DAMD 17-01-10707 and the Hayward Foundation (MRW) and DAMD 17-03-1-0224 (DM). We thank Debbie Neubauer, Elizabeth Baldwin and Trevor Lewis for their assistance in performing the experiments, Dr Lian Zhang and Frederick Kweh for mouse breeding and colony maintenance, and the University of Florida Cytogenetics Lab for cytogenetic analysis. All MRI data were obtained at the Advanced Magnetic Resonance Imaging and Spectroscopy (AMRIS) facility in the McKnight Brain Institute and National High Magnetic Field Laboratory at the University of Florida.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Perrin, G., Li, H., Fishbein, L. et al. An orthotopic xenograft model of intraneural NF1 MPNST suggests a potential association between steroid hormones and tumor cell proliferation. Lab Invest 87, 1092–1102 (2007). https://doi.org/10.1038/labinvest.3700675
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700675
Keywords
This article is cited by
-
Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966
Orphanet Journal of Rare Diseases (2020)
-
Immortalization of human normal and NF1 neurofibroma Schwann cells
Laboratory Investigation (2016)
-
MicroRNA profiling of peripheral nerve sheath tumours identifies miR-29c as a tumour suppressor gene involved in tumour progression
British Journal of Cancer (2013)
-
Mortality Associated with Neurofibromatosis 1: A Cohort Study of 1895 Patients in 1980-2006 in France
Orphanet Journal of Rare Diseases (2011)
