
Abstract
The administration of tumor necrosis factor-α (TNF-α) or the anti-Fas antibody (Jo-2) to mice causes acute liver failure, which is lethal within hours as a result of the induction of apoptosis in hepatocytes. It was recently reported that nonobese diabetic (NOD) mice are less sensitive to TNF-α/D-galactosamine (GalN)-induced liver failure than C57BL/6J (B6) mice, whereas both NOD and B6 mice were sensitive to the lethal effect of Jo-2. In the present study, we investigated the differences between the apoptotic liver cell death induced by TNF-α/GalN and that induced by Jo-2. B6, NOD, and Jcl-Imperial Cancer Research (ICR) mice were injected intravenously with TNF-α/GalN or Jo-2. ICR mice were less sensitive to TNF-α/GalN-induced liver failure than NOD and B6 mice (P<0.0001). In contrast, ICR mice were more sensitive to Jo-2-induced liver failure than B6 mice (P=0.0003). The liver caspase-3, -8 activity, serum transaminase levels, and the number of apoptotic liver nuclei all decreased in ICR in comparison to B6 mice treated with TNF-α/GalN. The mRNA expression of TNFR-associated death domain, Fas associated protein with death domain, and Bcl family and nuclear factor-κB activation induced by TNF-α/GalN were similar in both mice. Interestingly, the short form of cellular FLICE/caspase-8-inhibitory protein (c-FLIPS) was constitutively upregulated in ICR mice. In conclusion, these results suggest that ICR mice have an intrinsic resistance to TNF-α-induced hepatocyte apoptosis, and that c-FLIPS may play a role in TNF-α/GalN-induced liver failure, but not in Fas-induced liver failure.
Similar content being viewed by others


Main
Tumor necrosis factor-α (TNF-α) and the Fas ligand are cell-death mediators that act by binding to their responsive receptors, two TNF receptors (TNFR1 and TNFR2), and Fas respectively, and induce apoptosis in a variety of cell types. Both TNFR1 and Fas share a related C-terminal intracellular domain referred to as the ‘death domain’ and use similar types of associated molecules in the signal transduction pathway leading to apoptosis.1, 2, 3 The administration of TNF-α or the anti-Fas antibody (Jo-2) to mice causes acute liver failure that is lethal within hours as a result of the induction of apoptosis in hepatocytes.4, 5, 6 Unlike Fas related apoptosis, TNFR-mediated apoptosis requires hepatocyte sensitization by D-galactosamine (GalN).7, 8 The increased susceptibility of rodents to TNF-α-induced death after pretreatment with GalN is mainly caused by relatively selective liver failure. It was demonstrated that transcriptional inhibition by GalN in the mouse liver increases the sensitivity to the lethal effects of TNF-α thousands-fold.9 GalN may selectively block transcription in hepatocytes by depleting uridine nucleotides necessary for the production of mRNA transcripts.10 Tumor cells and cell lines display varied sensitivity to TNF; some cell lines are readily lysed by low concentrations, whereas others appear to be resistant even at high TNF concentrations. Moreover, some human cell lines are sensitive to Fas-mediated cytotoxicity but not to that induced by TNF-α, although they express both Fas and TNFR1.5, 11 Therefore, whether they trigger cell death along the same or distinct pathway in liver injury is still unclear. The interaction of TNF-α and its receptor leads in almost all cell types to activation of caspase-8, c-jun N-terminal kinase (JNK), and/or nuclear factor-κB (NF-κB).12 NF-κB promotes survival, whereas caspase-8 and JNK enhance cell death. NF-κB regulates the expression of anti-apoptotic gene products, including cellular FLICE/caspase-8-inhibitory protein (c-FLIP), the inhibitor of apoptosis proteins, Bcl-2, and Bcl-XL.13, 14, 15 Thus, NF-κB may negatively regulate TNF-α-induced apoptosis through regulation of c-FLIP levels, and its activation serves as a check point that determines whether TNFR will signal cell death or not.
c-FLIP is a human homolog of viral FLIP, which is a component of c-herpes viruses and has anti-apoptotic activity.16 c-FLIP molecules have several alternative splicing variants at transcriptional level, but at protein level two isoforms, long form of c-FLIP (c-FLIPL) and short form of c-FLIP (c-FLIPS), exist.17, 18, 19 The isoforms commonly contain two death effecter domains (DED), which resemble the N-terminal half of caspase-8 and serve as the binding motif for interaction with Fas associated protein with death domain (FADD).17, 19, 20 Owing to its structural homology with caspase-8, c-FLIP interferes with the activation of caspase-8 at the level of the death inducing signaling complex (DISC). c-FLIPS act as dominant-negative inhibitors of caspase-8 by preventing the processing and release of active caspase-8 from the receptor.19
A recent study by Bahjat et al21 demonstrated that the nonobese diabetic (NOD) mice are resistant to doses of TNF-α and GalN (TNF-α/GalN) that uniformly cause lethality in C57BL/6J (B6) mice, in contrast, no differences were seen between NOD and B6 mice in responsiveness to Jo-2. NOD mouse is a well-known inbred (genetically homogeneous) animal model of autoimmune type I diabetes derived from a closed colony (genetically heterogeneous) of Jcl-Imperial Cancer Research (ICR) mice.22 In addition, ICR mouse is closed colony mouse, which is originated from albino Swiss mouse. Japan SLC brought them from Charles River of USA in 1965, since then they have bred and provided in Japan. ICR mouse is usually used in the pharmacological test of the drug safety and effectiveness. Bahjat et al21 compared the lethality and liver cell death between NOD mice and B6 mice, not ICR mice, whereas the underlying mechanisms on the differential susceptibility of mice to TNF-α and GalN-mediated hepatocellular apoptosis remain unresolved.
In the present study, we demonstrated that ICR mice were resistant to the lethality induced by TNF-α/GalN, whereas neither B6 nor NOD mice showed such resistance. These findings led us to hypothesize that genetic determinants of TNF-α-induced hepatocyte apoptosis and lethality may be present in ICR mice, but not in NOD mice. We further showed that resistance to the hepatocyte apoptosis signaling pathway induced by TNF-α correlates with constitutive high expression of c-FLIPS, an inhibitor of caspase-8.
Materials and methods
Mice
Female B6, NOD, ICR, C3H/He, and BALB/c mice between 6 and 8 weeks of age were purchased from Japan SLC (Shizuoka, Japan). They were kept for at least 1 week under environmentally controlled conditions, with free access to food and water.
Reagents
GalN hydrochloride was purchased from Nacalai Tesque (Tokyo, Japan). Recombinant mouse TNF-α (rmTNF-α) was obtained from Genzyme (Cambrigde, MA, USA). Monoclonal hamster Jo-2 was purchased from Pharmingen (San Diego, CA, USA). For administration in mice, all materials were dissolved in pyrogen-free phosphate-buffered saline (PBS) (Gibco Invitrogen Corporation, Carlsbad, CA, USA). Antibodies against procaspases-3, -8, Bid, FLIPL/S, FADD, and actin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against JNK and phospho-specific-JNK were from Cell Signaling Inc. (Beverly, MA, USA). The anti-rabbit IgG horseradish peroxidase-coupled secondary antibody and ECL Western blot detection system were obtained from Amersham-Pharmatica Biotech (Buckinghamshire, UK).
Experimental Treatment
The experiments were conducted in accordance with the institutional guidelines by Gifu University School of Medicine. GalN was dissolved in PBS and was administered to mice intravenously (0.38 mg/g BW (bodyweight). rmTNF-α (24 ng/g BW) and Jo-2 (0.3 μg/g BW) was diluted with PBS to the required concentrations and injected to mice intravenously. The mice were killed at indicated time points, their livers were perfused via inferior vena cava with 10 ml of PBS and they were harvested for histological and chemical analyses, or they were snap-frozen in liquid nitrogen and stored at −80°C for subsequent molecular analyses.
Biochemical Analysis
Serum alanine transaminase (ALT) was measured using a standard clinical automatic analyzer.
Histologic Studies and Terminal Deoxynucleotidyl Transferase (TdT) Nick End-Labeling Assay
The liver was excised, fixed with 10% buffered formalin, sectioned at a thickness of 5 μm, and stained with hematoxylin and eosin for light-microscopic examination. Apoptotic cells were estimated using the terminal transferase dUTP nick end labeling (TUNEL) assay, which relies on incorporation of labeled dUTP at sites of DNA breaks. For the TUNEL procedure, all reagents, including buffers, were parts of a kit (Apop Tag; Oncor, Gaithersburg, MD, USA). All procedures were carried out according to the manufacturer's instructions. Apoptosis was examined in 5-μm-thick sections of the liver obtained from mice at indicated time points after TNF-α/GalN injection.
Caspase Activity
Frozen liver tissue was homogenized in 10 mmol/l HEPES buffer (pH 7.4) containing 2 mmol/l ethylenediaminetetraacetic acid, 0.1% 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate, 5 mmol/l dithiothreitol, 1 mmol/l phenylmethylsulfonyl fluoride, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, and 20 μg/ml leupeptin. After centrifugation at 14 000 r.p.m. at 4°C for 20 min, the supernatant (80 μg protein) was assayed for caspase activity using synthetic substrates: Ac-DEVD-pNA (Ac-Asp-Glu-Val-Asp-p-Nitroanilide) (Alexis, San Diego, CA, USA) for caspase-3 and Ac-IETD-pNA (Ac-Ile-Glu-Thr-Asp-pNa) (Oncogene Research Products, Cambridge, MA, USA) for caspase-8 at concentrations of 200 μmol/l. The kinetics of the proteolytic cleavage of the substrates was monitored in a microplatemeter (NJ-2300, System Instruments, Tokyo, Japan) using a 412-nm wavelength.
Western Blot Analysis
Frozen liver tissue specimens were homogenized in 1 ml ice-cold lysis buffer (50 mmol/l Tris-HCl, pH 7.4, 5 mmol/l EGTA, 1 mmol/l EDTA, 0.3 mmol/l PMSF, 30 μg/ml E64) by 20 strokes of a Potter-type homogenizer. The same volume of RIPA buffer (50 mmol/l Tris-HCl, pH 8.8, 150 mmol/l NaCl, 10 mmol/l EGTA, 1% Triton-X, 0.1% sodium dodecyl sulfate (SDS), 1% deoxycholic acid, and 0.3 mmol/l PMSF, 30 μg/ml E64) was added to the homogenate and the samples were then sonicated. Extracted proteins (50 μg) from frozen tissues were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and were electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were probed with the antibodies against procaspases-3, -8, Bid, FLIPL/S, FADD, JNK, phospho-specific-JNK, or actin, and then incubated with the anti-rabbit IgG horseradish peroxidase-coupled secondary antibody. Peroxidase activity on the PVDF membranes was visualized on X-ray film by means of the ECL Western blotting detection system. The density of each band was quantified by image analyzer using NIH Image J 1.34s software on a Macintosh computer system.
RNA Extraction and Ribonuclease Protection Assay
For the ribonuclease protection assay (RPA), the frozen liver was homogenized in ISOGEN (Nippongene, Tokyo, Japan), and total RNA was isolated as described previously.23 RPA was performed with the RiboQuant Multi-Probe Rnase Protection Assay System (Pharmingen) according to the manufacturer's instructions. Total RNA was hybridized with 32P-labeled probes including cytokine genes synthesized by mAPO-2 and mAPO-3 Multi-Probe Template sets (Pharmingen). After hybridization, the samples were treated with ribonuclease and then were electrophoresed on 5% polyacrylamide gels with 0.5 × Tris-borate-ethylenediaminetetraacetic acid buffer.
Nuclear Extracts and Electrophoretic Mobility Shift Assay
Nuclear extract of the livers were prepared as described previously.24 Nuclear extracts were frozen and stored at −80°C until use. Protein concentrations were measured by Bradford method. A double-stranded NF-κB consensus oligonucleotide probe (5′-TAGTTGAGGGGACTTTCCCAGGCA-3′) was labeled with [α-32P] deoxycytidine 5′-triphosphate (ICN Biomedical, Costa Mesa, CA, USA) using Klenow DNA polymerase. Protein–DNA binding reactions were performed for 30 min at room temperature using 20 μg of nuclear protein and 2.5 ng of 32P-labeled double-stranded oligonucleotide probe. For competition reactions, unlabeled oligonucleotide was added 10 min before the addition of the radiolabeled probe. The mixture was electrophoresed through 4% polyacrylamide Tris-borate-ethylenediaminetetraacetic acid gels.
Statistical Analysis
The differences in survival between ICR, NOD, B6, C3H, and BALB/c mice were determined by Kaplan–Meier analyses or χ2-test. The Kaplan–Meier curves were statistically compared by the log-rank-test. All values in the figures are expressed as the mean±s.e. The significance of differences among mean values was evaluated according to the Mann–Whitney U-test.
Results
TNF-α/GalN-Induced Lethality in NOD, B6, and ICR Mice
NOD, B6, and ICR mice were injected intravenously with TNF-α/GalN. The mice were monitored for up to 12 h, and the percent survival was determined. B6 mice started to die within 4 h after TNF-α/GalN injection, and the final mortality was 100%. NOD mice started to die within 5 h after TNF-α/GalN injection and the mortality at 12 h was 50%. In contrast, all ICR mice survived, indicating their resistance to TNF-α/GalN-induced liver failure (Figure 1a) (P<0.0001 for overall comparison). Although NOD was less sensitive to TNF-α/GalN than B6 (P<0.0001) as reported previously,21 its genetic background ICR was more resistant (P<0.0001 as compared to B6) (Figure 1a). According to these results, we decided to compare B6 and ICR mice to investigate their intrinsic sensitivity to TNF-α/GalN-induced liver failure.

Lethality in TNF-α/GalN or Fas agonist treated mice. (a) Kaplan–Meier survival curves in TNF-α/GalN treated B6, NOD, and ICR mice. Mice were challenged intravenously with GalN (0.38 mg/g BW) and rmTNF-α (0.024 μg/g BW). The mice were monitored for up to 12 h, and the percent survival was determined. The overall difference in the survival among ICR, NOD, and B6 mice were highly significant (P<0.0001 by the log-rank test). (b) Kaplan–Meier survival curves in Fas agonist (Jo-2) treated B6 and ICR mice. Mice were challenged with Jo-2 (0.3 μg/g BW). The mice were monitored for up to 4 h, and percent survival was determined. The difference in the survival between ICR and B6 mice were highly significant (P=0.0003 by the log-rank test).
Jo-2-Induced Lethality in B6 and ICR Mice
Because the TNFR1 shares with Fas an intracellular signaling pathway that converges at the level of FADD and caspase-8, we next examined the responsiveness of ICR mice to a Fas agonist. ICR and B6 mice were injected intravenously with Jo-2. The mice were monitored for up to 4 h, and percent survival was determined. Although ICR mice were more sensitive to Jo-2-induced liver failure than B6 mice (P=0.0003), both strains died within 4 h (Figure 1b).
Liver Injury and Apoptosis in TNF-α/GalN-Treated B6 and ICR Mice
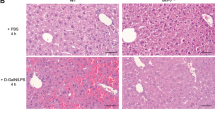
The mice treated with TNF-α/GalN were killed at the indicated time points as shown in Figure 2a. Serum ALT activities rose in B6 mice from 3 h after TNF-α/GalN injection up to 6 h. On the other hand, no elevation in the serum ALT, activities was observed in the ICR mice (Figure 2a, P<0.001 as compared to B6). TUNEL assay revealed that hepatocyte apoptosis was suppressed in the ICR mice in comparison to B6 (Figure 3).

Liver injury in TNF-α/ GalN or Fas agonist treated mice. (a) Three mice per group were challenged intravenously with GalN (0.38 mg/g BW) and rmTNF-α (0.024 μg/g BW). The mean sALT activity measured at the time of autopsy is indicated for each group and is expressed as international units per liter (mean±s.e.). (b) Three mice per group were injected intravenously with Jo-2 (0.3 μg/g BW). The mean sALT activity measured at the time of autopsy is indicated for each group and expressed as international units per liter (mean±s.e.). *P<0.05, **P<0.01 in comparison to the value at the same indicated time point of ICR mice.

Induction of apoptosis in the liver of TNF-α/GalN treated mice. B6 mice (a and c) and ICR mice (b and d) were challenged intravenously with GalN (0.38 mg/g BW) and rmTNF-α (0.024 μg/g BW) and killed 2 h (a and b) and 4 h (c and d) after injection. Liver specimen was subjected to an in situ TUNEL assay to detect apoptotic cells (original magnification, × 100).
To assess whether caspases are involved in the apoptotic process induced by TNF-α/GalN injection, the cleavage of pro-caspases-3, -8, and Bid was examined by a Western blot analysis (Figure 4a). The active form of caspase-8 is generated upon proteolytic cleavage of pro-caspase-8 by DISC and is able to cleave and activate downstream caspases such as caspase-3. The pro-capase-8 level decreased at 4 h after administration, and thereafter it continued to decrease until 6 h in B6 mice. Pro-caspase-3, which ultimately elicits the morphological hallmark of apoptosis such as DNA fragmentation and cell shrinkage, decreased at 4 h after administration, and also continued to decrease until 6 h in B6 mice. On the other hand, pro-caspase-8 and pro-caspase-3 did not decrease until 6 h after administration in ICR mice. The active form of bid is generated after proteolytic cleavage by caspase-8 and then it is translocated to the mitochondria where it induces cytochrome c release and mitochondria damage.25 Bid also decreased in B6 mice although its level in the liver of ICR mice did not change. Consistent with a Western blot analysis, elevations in the caspase-8-like activity and capsase-3-like activity were observed in B6 mice from 2 to 6 h after TNF-α/GalN injection (Figure 4b. Fivefold increases, and 17-fold increases over the control level, respectively). Although both caspase-8-like activity and caspase-3-like activity increased in ICR mice, these levels were significantly lower than those in the livers from B6 mice (Figure 4b, P<0.05).

Activation of caspases and Bid after TNF-α/GalN injection. (a) Total liver proteins were isolated from the livers of TNF-α/GalN treated mice at the indicated time points. Proteins (50 μg) were subjected to SDS-PAGE, and were probed with specific antibodies against procaspase-8, -3, Bid, and actin. The results shown are representative of at least three independent experiments. The histogram shows quantitative representations of the levels of pro-caspase-8, Bid, and pro-caspase-3 obtained from laser densitometric analysis. Data are the mean±s.e. from at least three independent experiments. *P<0.05 in comparison to the value at 0 h of B6 mice. (b) The caspase activities were monitored by the cleavage of peptide substrates. For the measurement of caspase-3 (like) and -8 (like) protease activities, the substrates Ac-DEVD-pNA and Ac-IETD-pNA were used, respectively. The mean caspase activities measured at the time of autopsy are indicated for each group and expressed by fold increase to those of control (0 h). The data are the mean±s.e. from at least three independent experiments. *P<0.05 in comparison to the value at the same indicated time point of B6 mice.
Liver Injury and Apoptosis in Jo-2-Treated B6 and ICR Mice
Liver injury as assessed by serum ALT activities was observed in ICR mice 1 h after Jo-2 injection, and reached its peak at 2 h. Although serum ALT activities of B6 mice at 2 h after injection were significantly lower than those of ICR mice (Figure 2b, P<0.05), the peak serum ALT activities of B6 mice at 3 h were similar to those of ICR mice at 2 h after Jo-2 injection. The levels of pro-caspase-8 and pro-caspase-3 comparably decreased in the ICR mice and B6 mice after Jo-2 injection (Figure 5a) although caspase-3-like activity of ICR mice at 0.5 h was significantly higher than that of the B6 mice (Figure 5b, P<0.05). On the basis of these results, we consider that ICR mice are resistant only against TNFR-mediated hepatocyte apoptosis but not against Fas-mediated apoptosis.

Activation of caspases and Bid after Fas agonist injection. (a) Total liver proteins were isolated from livers of Jo-2 treated mice at indicated time points. Proteins (50 μg) were subjected to SDS-PAGE, and were probed with specific antibodies against procaspase-8, -3, Bid, and actin. The results shown are representative of at least three independent experiments. (b) Caspase activities were monitored by the cleavage of peptide substrates. For the measurement of caspase-3 (like) and -8 (like) protease activities, the substrates Ac-DEVD-pNA and Ac-IETD-pNA were used, respectively. The mean caspase activities measured at the time of autopsy are indicated for each group and expressed by fold increase to those of control (0 h). Data are the mean±s.e. from at least three independent experiments. *P<0.05 in comparison to the value at the same indicated time point of B6 mice.
Involvement of JNK-Dependent Apoptotic Pathway in TNF-α/GalN- or Jo-2-Treated B6 and ICR Mice
As it is recognized that the binding of TNF-α or Fas to its receptor leads to activation of caspase-8 and/or JNK,12 we examined the levels of JNK phosphorylation after TNF-α/GalN or Jo-2 injection in B6 and ICR mice. As a result, JNK phosphorylation was observed in B6 mice but not ICR mice after TNF-α/GalN injection. In contrast, JNK phosphorylation was not observed either in B6 nor ICR mice after Jo-2 injection (data not shown). These results suggest that JNK-dependent apoptotic pathway is not involved in Jo-2-induced liver failure in these mice.
mRNA and Protein Expression of Pro- and Anti-Apoptotic Molecules
One possible explanation for the different response between ICR and B6 mice may be the inability of rmTNF-α to bind to the TNFR1 receptor on ICR cells. We first performed RPA to evaluate the TNFR1 mRNA expression in the liver. The mRNA expressions of TNFR1 (TNFRp55) were similar in ICR and B6 mice, and they did not change after TNF-α/GalN injection. In addition, the expressions of FADD and TNFR-associated death domain (TRADD) mRNA, which form the DISC, were the same levels, and did not change after TNF-α/GalN injection (Figure 6a). By a Western blot analysis, FADD levels appeared to be consistent between both mice during the time course examined (Figure 6b). As, caspase-3 like activity after TNF-α/GalN injection was slightly elevated even in ICR mice, another possible explanation for the different response between ICR and B6 mice may be owing to an upregulation of anti-apoptotic genes or downregulation of pro-apoptotic genes in the liver of ICR mice. We performed RPA to evaluate the Bcl family mRNA expression obtained from the liver tissues after TNF-α/GalN injection. There was no difference in mRNA expression levels of anti-apoptotic genes such as Bcl-XL, Bfl-1 and Bcl-2, and pro-apoptotic genes such as Bak, Bax, and Bad, between ICR and B6 mice 2 and 4 h after TNF-α/GalN injection (Figure 6c).

mRNA and protein expressions of DISC forming molecules, and mRNA expressions of pro- and anti-apoptotic genes. (a) Total hepatic RNA (20 μg) was isolated from livers of TNF-α/GalN treated mice at indicated time points and analyzed for FADD, TRADD, and TNFR1 mRNA expressions by RPA. (b) Total proteins were isolated from livers of TNF-α/GalN treated mice at indicated time points. Proteins (50 μg) were subjected to SDS-PAGE, and were probed with specific antibodies against FADD and actin. The results shown are representative of at least three independent experiments. (c) Total hepatic RNA (20 μg) was isolated from the livers at the indicated time points and then was analyzed for Bcl family mRNA expression by RPA.
Activation of NF-κB
The cytotoxicity mediated by TNFR1 is regulated by two kinds of opposing activities: induction of apoptosis and activation of the transcription factor, NF-κB which increase ‘survival genes’ and act as anti-apoptosis. We next investigated whether NF-κB activation can reveal any difference after TNF-α/GalN injection in the liver between ICR mice and B6 mice. Nuclear NF-κB activation was induced comparably in B6 and ICR mice (Figure 7). These results suggest that survival signals through TNFR1-NF-κB pathway was similar and functional in hepatocytes of ICR and B6 mice. Taking these findings into account as a whole, the ability of TNF-α to bind to the TNFR1 receptor on hepatocytes of ICR mice is thus considered to be similar to that of B6 mice.

NF-κB DNA-binding activity in the liver after TNF-α/GalN treatment in B6 and ICR mice. Nuclear extract was isolated from the livers at the indicated time points and 20 μg of nuclear protein was analyzed by electrophoretic mobility shift assay. The results shown are representative of at least three independent experiments. NS; non-specific band.
Differential Expression of FLIPS
Next, we hypothesized that a difference in the susceptibility to TNF-α/GalN-induced hepatocyte apoptosis may exist upstream of the caspase-8 activation. We paid attention to c-FLIP, an inhibitor of caspase-8. To assess whether c-FLIPL and c-FLIPS protein levels were upregulated in ICR mice, B6, NOD, and ICR mice were killed and protein obtained from liver tissue was analyzed by a Western blot analysis. The levels of c-FLIPL showed no difference among these mice until 4 h after TNF-α/GalN injection. In contrast, the levels of c-FLIPS in ICR mice were upregulated constitutively (see Figure 8a, 0 h) and remained high after TNF-α/GalN injection as compared to that in B6 mice (Figure 8a). In addition, the expression levels of c-FLIPS in NOD mice were constitutively higher than those of B6 mice, but lower than those of ICR mice. Next, to clarify whether differential c-FLIPS expression accounts for the strain difference, we examined the relationship between the lethality of mice to TNF-α/GalN-induced liver failure and the expression levels of c-FLIPS among different strains of mice. In addition to B6 and ICR mice, the levels of c-FLIPS in the livers and TNF-α/GalN-induced lethality were determined also in NOD, C3H, and BALB/c mice. As shown in Figure 8b, ICR, and BALB/c mice were resistant to TNF-α/GalN-induced lethality, whose survival rates were 100 and 70%, respectively. In contrast, all B6 and C3H mice died after TNF-α/GalN were administrated. Constitutive expression levels of c-FLIPS, but not c-FLIPL, in the liver of ICR and BALB/c mice were significantly higher than those in B6 and C3H mice (Figure 8b). These results suggest that expression levels of c-FLIPS correlated with the survival rates of mice treated with TNF-α/GalN.

Differential expression of c-FLIPs. (a) Time course of the c-FLIP expression after TNF-α/GalN injection in B6, NOD, and ICR mice. Total proteins were isolated from the livers of the TNF-α/GalN treated mice at the indicated time points. Proteins (50 μg) were subjected to SDS-PAGE, and were probed with specific antibodies against c-FLIPL/S and actin. The results shown are representative of at least three independent experiments. (b) The relationship between the lethality of mice to TNF-α/GalN-induced liver failure and the expression levels of c-FLIPs among different strains of mice. Mice were challenged intravenously with GalN (0.38 mg/g BW) and rmTNF-α (0.024 μg/g BW; upper). The mice were monitored for up to 12 h, and the percent survival was determined. *P<0.05 and **P<0.0001 in comparison to B6 mice by the χ2-test (middle). Total proteins were isolated from the livers of TNF-α/GalN untreated mice from B6, ICR, C3H, NOD, and BALB/c strain. Proteins (50 μg) were subjected to SDS-PAGE, and were probed with specific antibodies against c-FLIPL/S and actin. The results shown are representative of at least three independent experiments (lower). The histogram shows quantitative representations of the levels of c-FLIPS obtained from laser densitometric analysis. Data are the mean±s.e. from at least three independent experiments. *P<0.05 in comparison to the value of B6 mice.
Discussion
Our results show that ICR mice are less sensitive to TNF-α/GalN-induced hepatocyte apoptosis than B6 mice. This event correlates with the finding that the ICR mice have constitutively upregulated FLIPS expression in the liver, because the constitutive expression levels for c-FLIPS in the liver were different among various mouse strains including B6, ICR, NOD, C3H, and BALB/c mice, and these levels were associated with the survival rates of mice treated with TNF-α/GalN. We would propose a possible mechanism to explain the resistance of ICR mice: (1) constitutive upregulation of c-FLIPS in ICR mice, probably because of genetic background; (2) inhibition of pro-caspase-8 cleavage by c-FLIPS in the DISC; (3) reduced apoptotic signals leading to the activation of pro-caspase-3; and (4) resistance to the lethal effect of TNF-α/GalN injection.
Soluble or membrane-bound cytokines such as TNF-α or Fas ligand play a prominent role in immunologic, inflammatory, and pathologic conditions in a variety of diseases including hepatitis. Although the signaling pathway through TNFR1 is similar to that activated by Fas, TNFR1 signaling differs from Fas pathway in several steps as follows. First, binding of FADD and RIP to the receptor complex requires the adaptor protein TRADD.26 Second, binding of Daxx to TNFR1 has not yet been shown and the ASK1–JNK apoptotic pathway is activated by ROS.27, 28, 29
In a series of studies, it has been demonstrated that there are significant variations in lipoplysaccharide and GalN-induced lethality among different strains of mice. The host immune response, and particularly the degree of TNF-α production, can modulate the liver injury, and transgenic mice expressing null forms of TNF-α, TNFR1, or expressing only a cell-associated forms of TNF-α exhibited no mortality and only a modest degree of liver injury.30 However, there have so far been few studies addressing that a resistance to TNF-α-associated lethality and apoptosis is due to a post-receptor step in TNFR1 signaling. Bahjat et al21 observed that NOD mice were resistant to the lethal effects of TNF-α/GalN but responsive to those of Jo-2. However, the defect seen in the TNFR1 signaling pathway of NOD mice was only partly elucidated although they suggested that the defect appeared to be a post receptor event, because the binding of recombinant human TNF-α to NOD splenocytes appeared to be normal. NOD mice have specific genetic host factors, particularly those related to immune modulation that may be important in determining the differential responses to injury from ICR mice, thus resulting in the development of type I diabetes whereas ICR mice did not.31 The present data in our study clearly show that the defect seen in TNFR-mediated apoptotic signaling pathway is not associated with the specific autoimmune predisposition of NOD mice, which may cause type I diabetes, but is associated with genetic host factor(s) of background ICR mice.
Consistent with Bahjat et al's study, our data indicate that NOD mice do not lack functional TNFR1 on their hepatocytes, because TNFR1 mRNA, TRADD mRNA, and FADD mRNA, and FADD were comparably detected both in ICR and B6 (Figure 6), and caspase-8 and -3 activities were markedly reduced, but not absent in ICR mice. An alternative explanation for the reduced apoptotic cell death in hepatocytes from ICR mice is that NF-κB activation is increased in ICR mice, because NF-κB plays a pivotal role in the crossroads between life and death in a variety of cells including hepatocytes.24, 32 However, NF-κB was similarly induced either in ICR mice or B6 mice after TNF-α/GalN injection (Figure 7), indicating that NF-κB-related anti-apoptotic signaling is equally induced in both mice, as well as that TNF-α induces NF-κB activation through TNFR1. It is well known that the adapter protein TRADD binds to TNFR1 and serves as anchor for the subsequent recruitment of other proteins into the signaling complex that directly lead to cell death or NF-κB induction. These results suggest that the ability of TNFR1 to form a signaling complex with TRADD and FADD has no difference between ICR and B6 mice.
The studies on caspase activation appear to demonstrate that the inhibitory effect observed in ICR mice on TNFR-mediated hepatocyte apoptosis was upstream of caspase-8 activation, because TNF-α-induced caspase-8 activation in the liver was inhibited in ICR mice compared with B6 mice (Figure 4). These findings led us to determine anti-apoptotic protein c-FLIP, an inhibitor of caspase-8. Both c-FLIPS and c-FLIPL commonly contain two DED, which resemble the N-terminal half of caspase-8 and serve as the binding motif for interaction with FADD.17, 20 c-FLIPL additionally contains a caspase-like domain at its C-terminal part that is deficient in c-FLIPS. However, the functional differences between these variants are still not fully understood. c-FLIPS have been reported to completely block the initial cleavage step of pro-caspase-8 at DISC in its activation processing, whereas c-FLIPL allows the initial cleavage step, thus generating the p10 subunit, but blocking any further processing.33 In addition, the recruitment of c-FLIPL into FADD occurs later than that of c-FLIPS.34 In the present study, the expression levels of c-FLIPS in ICR mice was constitutively upregulated, whereas the expression levels of c-FLIPL until 2 h after TNF-α/GalN injection was similar. Because the caspase-8-like activity was significantly higher in B6 mice 2 h after TNF-α/GalN injection, upregulated c-FLIPS in ICR mice may explain why ICR mice are less sensitive to TNF-α/GalN-induced liver injury. Although a number of studies that c-FLIPS plays a pivotal role in the inhibition of caspase-8 dependent cell death in vitro are reported,35 there is only one report about in vivo overexpression of c-FLIPS in thymocytes and in mature T cells,36 in addition, c-FLIP-deficient animals have been reported not to survive past day 10.5 of embryogenesis.35 These situations make it difficult for us to confirm the role of c-FLIPS in the resistance of hepatocyte apoptosis in ICR mice. On the contrary, there are many reports about in vitro models of c-FLIPS overexpression, which indicates the anti-apoptotic property of c-FLIPS.19, 20 Moreover, we found the correlation between the expression levels of c-FLIPS and the survival rates of mice treated with TNF-α/GalN among different strains of mice. These findings support a role of c-FLIPS in susceptibility to TNF-α-induced apoptosis. Akt is well known to regulate FLIP expression.37 It has been reported that c-FLIPS and c-FLIPL are differently regulated,38 although the difference in the regulatory mechanisms remains unclear. Genetic host factors may be important in determining differential expressions of c-FLIPS among various strains of mice. Further studies will be needed to elucidate the differences.
The explanation for opposing lethality in ICR and B6 mice treated with Jo-2 was not elucidated, as c-FLIPS have been reported to inhibit not only TNF-α-induced but also Fas-induced FADD-mediated apoptosis in a variety of cell types.19 In addition, some human cell lines have been reported to be sensitive to Fas-mediated cytotoxicity but not to that induced by TNF-α, although they express both Fas and TNFRl.11 Fas-derived signals were also reported to have the most rapid effect, killing most cells in a human melanoma line within hours of stimulation, whereas TNFR1-associated signals required 2–3 days after ligand binding for cell death to occur.39 These findings suggest that TNFR and Fas transduce distinct signal(s), thus resulting in apoptosis. In addition, Fas has been reported to bind Daxx and induce apoptosis by activating the ASK1–JNK apoptotic pathway, although the binding of Daxx to TNFR1 has not been shown.27, 28, 29 However, in the present study, the ASK1–JNK apoptotic pathway was not involved in Jo-2-induced liver failure in both mice. Taking together, unlike TNFR1-mediated hepatic apoptosis, it is likely that the caspase-8 independent signaling pathway leading to caspase-3 activation may be functional in Fas-mediated liver cell death. Further investigation is required to address such interesting and important question. Alternatively, as in type I cells (mitochondria-independent), death receptors induce strong caspase-8 activation, which alone is sufficient to lead to robust processing of effector caspases and apoptosis induction, but in type II cells (mitochondria-dependent), only low amounts of caspase are activated on DISC formation,40 upregulation of c-FLIPS observed in the livers of ICR mice may be insufficient to inhibit a strong caspase-8 activation in the mitochondria-independent apoptotic pathway mediated by Fas. We have recently demonstrated that the mitochondria-independent pathway can compensate Fas-mediated hepatic apoptosis but not TNFR-mediated hepatic apoptosis.41
To the best of our knowledge, the present study is the first to show that the expression level of c-FLIPS was different on genetic background. Furthermore, our results suggest that c-FLIPS may have a role in TNF-α/GalN-induced liver cell injury. The observations that ICR mice are resistant to hepatocyte injury and lethality associated with TNFR1 receptor signaling and constitutive upregulation of c-FLIPS in ICR provide significant implications for the mechanisms of TNF-α and Fas ligand-induced apoptosis. Further studies are required to determine whether other types of cells from ICR mice are also resistant to TNF-α-mediated apoptosis.
References
Osthoff SK, Krammer PH, Droge W . Divergent signaling via APO-1/FAS and the TNF receptor, two homologous molecules involved in physiological cell death. EMBO J 1994;13:4587–4596.
Nagata S . Apoptosis by death factor. Cell 1997;88:355–365.
Liang H, Feski SW . Three-dimensional structures of proteins involved in programmed cell death. J Mol Biol 1997;274:291–302.
Gantner F, Leist M, Jilg S, et al. Tumor necrosis factor-induced hepatic DNA fragmentation as an early marker of T cell-dependent liver injury in mice. Gastroenterology 1995;109:166–176.
Leist M, Gantner F, Naumann H, et al. Tumor necrosis factor-induced apoptosis during the poisoning of mice with hepatotoxins. Gastroenterology 1997;112:923–934.
Ogasawara J, Watanabe-Fukunaga R, Adachi M, et al. Lethal effect of the anti-Fas antibody in mice. Nature 1993;364:806–809.
Nagaki M, Sugiyama A, Osawa Y, et al. Lethal hepatic apoptosis mediated by tumor necrosis factor receptor, unlike Fas-mediated apoptosis, requires hepatocyte sensitization in mice. J Hepatol 1999;31:997–1005.
Leist M, Gantner F, Kunstle G . The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocye apoptosis and subsequent liver failure. Mol Med 1996;2:109–124.
Galanos C, Freudenberg MA, Reutter W . Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci USA 1979;76:5939–5943.
Leist M, Gantner F, Jilg S, et al. Activation of 55 kDa TNF receptor is necessary and sufficient for TNF-induced liver failure, hepatocyte apoptosis, and nitrite release. J Immunol 1995;154:1307.
Wong GHW, Goeddel DV . Fas antigen and p55 TNF receptor signal apoptosis through distinct pathways. J Immunol 1994;152:1751–1755.
Wajant H, Pfizenmaier K, Scheurich P . Tumor necrosis factor signaling. Cell Death Differ 2003;10:45–65.
Wang CY, Mayo MW, Korneluk RG, et al. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and cIAP1 and cIAP2 to suppress caspase-8 activation. Science 1998;281:1680–1683.
Lee HH, Dadgostar H, Cheng O, et al. NF-κB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci USA 1999;96:9136–9141.
Micheau O, Lens S, Gaide O, et al. NF-κB signals induce the expression of c-FLIP. Mol Cell Biol 2001;21:5299–5305.
Thome M, Schneider P, Hofmann K, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997;386:517–521.
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997;388:190–195.
Scaffidi C, Schmitz I, Krammer PH, et al. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999;274:1541–1548.
Thome M, Tschopp J . Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol 2001;1:50–58.
Krueger A, Baumann S, Krammer PH, et al. FLICE-inhibitory proteins regulators of death receptor-mediated apoptosis. Mol Cell Biol 2001;21:8247–8254.
Bahjat FR, Dharnidharka VR, Fukuzuka K, et al. Reduced susceptibility of nonobese diabetic mice to TNF-α and D-galactosamine-mediated hepatocellular apoptosis and lethality. J Immunol 2001;165:6559–6567.
Makino S, Kunimoto K, Muraoka Y, et al. Breeding of non-obese, diabetic strain of mice. Exp Animals 1980;29:1–13.
Imose M, Nagaki M, Naiki T, et al. Inhibition of nuclear factor-κB and phosphatidylinositol 3-kinase/Akt is essential for massive hepatocyte apoptosis induced by tumor necrosis factor α in mice. Liver int 2003;23:386–396.
Nagaki M, Naiki T, Brenner DA, et al. Tumor necrosis factor α prevents tumor necrosis factor receptor-mediated mouse hepatocyte apoptosis, but not Fas-mediated apoptosis: role of nuclear factor-κB. Hepatology 2003;32:1272–1279.
Gross A, Yin XM, Wang K, et al. Caspase cleaved Bid targets mitochondria and is required for cytochrome c release, while Bcl-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem 1999;274:1156–1163.
Strasser A, O’Connor L, Dixit VM . Apoptosis signaling. Annu Rev Biochem 2000;69:217–245.
Yang X, Koshravi-Far R, Chang HY, et al. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 1997;89:1067–1076.
Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001;2:222–228.
Chang HY, Nishitoh H, Yang X, et al. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 1998;181:1860–1863.
Papadakis KA, Targan SR . Tumor necrosis factor: Biology and therapeutic inhibitors. Gastroenterology 2000;119:1148–1157.
Anderson MS, Bluestone JA . The NOD mice: a model of immune dysregulation. Annu Rev Immunol 2005;23:447–485.
Karin M, Lin A . NF-κB at the cross roads of life and death. Nat Immunol 2002;3:221–227.
Krueger A, Schmitz I, Baumann S, et al. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem 2001;276:20633–20640.
Park SJ, Kim YY, Ju JW, et al. Alternative splicing variants of c-FLIP transduce the differential signal through the Raf or TRAF2 in TNF-induced cell proliferation. Biochem Biophys Res Commun 2001;289:1205–1210.
Yeh WC, Itie A, Elia AJ, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000;12:633–642.
Oehme I, Neumann F, Bosser S, et al. Transgenic overexpression of the Caspase-8 inhibitor FLIP(short) leads to impaired T cell proliferation and increased memory T cell pool after staphylococcal enterotoxin B injection. Eur J Immunol 2005;35:1240–1249.
Nam SY, Jung GA, Hur GC, et al. Upregulation of FLIP(s) by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci 2003;94:1066–1073.
Bin L, Li X, Xu LG, et al. The short splice form of Casper/c-FLIP is a major cellular inhibitor of TRAIL-induced apoptosis. FEBS Lett 2002;510:37–40.
Clement MV, Stamenkovic I . Fas and tumor necrosis factor receptor-mediated cell death: similarities and distinctions. J Exp Med 1994;180:557–567.
Scaffidi C, Fulda S, Srinivasan A, et al. Two CD95 (AOP-1/Fas) signaling pathways. EMBO J 1998;17:1675–1687.
Imao M, Nagaki M, Imose M, et al. Differential caspase-9-dependent signaling pathway between tumor necrosis factor receptor-and Fas-mediated hepatocyte apoptosis in mice. Liver Int 2005;25:162–170.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takai, S., Nagaki, M., Imao, M. et al. Intrinsic resistance to TNF-α-induced hepatocyte apoptosis in ICR mice correlates with expression of a short form of c-FLIP. Lab Invest 87, 572–581 (2007). https://doi.org/10.1038/labinvest.3700544
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700544
