
Abstract
Recurrent chromosomal rearrangements have not been well characterized in common carcinomas. We describe the use of a novel bioinformatics approach to discover candidate oncogenic chromosomal aberrations on the basis of outlier gene expression called COPA (cancer outlier profile analysis). We demonstrate how this approach led to the identification of gene fusions of the 5′-untranslated region of TMPRSS2 (21q22.3), an androgen regulated gene, with the ETS transcription factor family members, either ERG (21q22.2), ETV1 (7p21.2), or ETV4(17q21). These novel gene fusions suggest a mechanism for overexpression of the ETS genes in the majority of prostate cancers identified through PSA screening. Considering the high incidence of prostate cancer and the high frequency of this gene fusion, the TMPRSS2-ETS gene fusions are the most common genetic aberration so far described in human malignancies. The clinical implications of this discovery are significant for diagnosis and potentially for the development of targeted therapy.
Similar content being viewed by others


Main
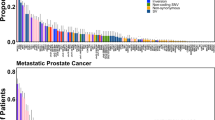
Microarray experiments generate copious data that can be used to identify significantly differentially expressed genes between known classes of samples. This approach can lead to the identification of molecular biomarkers. For example, AMACR (α-methylacyl CoA racemase), hepsin, and fatty acid synthetase are all over expressed in prostate cancer as compared to benign prostate tissue.1, 2, 3 Statistical significance for biomarkers is demonstrated by comparing the mean expression of one class to another. For example in Figure 1 (left, biomarkers profile), AMACR in prostate cancer (class 2, red) is significantly over expressed as compared to the reference class—benign prostate tissue (class 1, blue). These results are visually appreciated by ordering the expression of AMACR by class.

Cancer outlier profiler analysis (COPA). A cancer biomarker (left), such as AMACR, demonstrates significant over expression in the majority of cancer samples (red) as compared to benign samples (blue). An oncogene outlier profiler for ERG is characterized by significant over expression in a subpopulation of samples within the prostate cancer samples (red). Standard statistical tests such as the Student's t-test are useful for the biomarker profile but fail to identify profiles with only a few outlier cases. COPA transforms the data (as described in text) to accentuate profiles with outliers. These data are from the study by LaPointe et al.12
The difference in the mean AMACR expression between the two groups is statistically significant although there is some expression in benign tissues that is at a similar level to some prostate cancer samples. In order to rank the best biomarkers for a specific class, one can compare the results of multiple micorarray experiments in a meta-analysis approach. In a meta-analysis of four cDNA expression array data sets, AMACR was one of the genes most consistently over expressed in prostate cancer.4 This meta-analysis approach has lead to the development of the publicly available compendium of expression array data called Oncomine (www.oncomine.org) that allows researchers to investigate over 300 expression array data sets.5 However, one limitation to this standard biomarker analysis is how does it deal with genes significantly differentially expressed in only a subset of the tumors?
Tumor cells thrive by developing a growth advantage over neighboring benign cells through a variety of genetic and epigenetic alterations. Overexpression of oncogenes favors this growth advantage and can occur through gene copy number amplification, activating mutations or by constitutive promoter activation. Oncogenes such as her-2-neu or EGFR are examples where overexpression is observed in only a subset of tumors from patients with breast or lung cancer, respectively. Thus, the expression array profile of an oncogene, may look very different when compared to AMACR. In a recent study from our group, a simple approach was developed to identify oncogene profiles that can be characterized by overexpression of a small subset of biologically important outlier cases.
The method called cancer outlier profile analysis (COPA) was developed based on the idea that evaluating variance in a data set using the median instead of the mean would maintain the peaks of outliers. COPA has three steps. First, gene expression values are median centered, setting each gene's median expression value to zero. Second, the median absolute deviation (MAD) is calculated and scaled to 1 by dividing each gene expression value by its MAD (Figure 1). This approach was used instead of centering data around the mean because it has less effect on the tails or outliers. Third, the 75th, 90th, and 95th percentiles of the transformed expression values are tabulated for each gene and then genes are rank-ordered by their percentile scores, leading to a prioritized list of outlier profiles.
By applying COPA, 132 gene expression data sets representing 10 486 microarray experiments were interrogated for outlier genes.6 Examples of known genes that are over expressed in a subset of a particular tumor type were identified such as the oncogene her-2-neu and E-Cadherin (CDH1) (see Table 1). Interestingly, genes such as RUNX1T1 (ETO) and PBX1 also scored high on COPA. These two genes are known to be associated with the AML-ETO and E2A-PBX1 gene translocations in acute myeloid leukemia and acute lymphoblastic leukemia, respectively. Both of these translocations only occur in a subset of the cases (ie, outlier cases). Two genes consistently scored high in prostate cancer microarray experiments, ERG (Figure 1, right) and ETV1. Both of these genes are members of the ETS family of transcription factors. They were over expressed in the majority (50–70%) of prostate cancers and were mutually exclusive across several independent gene expression data sets, suggesting that they may be functionally redundant in prostate cancer development.6 As the ETS family of transcription factors has previously been seen in the genomic translocation of the Ewing's family tumors, AML and other rare tumors, the possibility that they were part of a translocation in prostate cancer was explored. When the ERG cDNA transcript was evaluated exon by exon, overexpression was seen at the distal (3′ end) but not the proximal portion (5′ end). By sequencing the cDNA transcripts, fusions of the 5′-untranslated region of TMPRSS2 (21q22.3) with the ETS transcription factor family members, either ERG (21q22.2), ETV1 (7p21.2),6 and more recently ETV420 were identified, suggesting a novel mechanism for overexpression of the ETS genes in prostate cancer (Figure 2).

Anatomy of the TMPRSS2 to ETS family gene fusions identified in prostate cancer. Adapted from Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644–648,6 reprinted with permission from AAAS.
Thus, the identification of these gene fusions between the prostate-specific, strongly androgen-regulated gene TMPRSS2 (21q22.3) to ERG, ETV1, or ETV4 was a surprising discovery. Using other methods to validate these findings (ie, RT-PCR and fluorescence in situ hybridization (FISH)) in human prostate cancer samples, the TMPRSS2:ETS gene fusions are seen in up to 80% of hospital-based clinical cohorts. TMPRSS2:ETS gene fusions have been detected in approximately 20% of the precursor lesion high-grade prostatic intraepithelial neoplasia (PIN) but not prostatic atrophy (PIA) (unpublished observations). As TMPRSS2 is regulated by androgens, even in the setting of hormone ablation therapy for metastatic prostate cancer, low levels of androgen may still be sufficient to drive ETS overexpression.
The TMPRSS2:ETS gene fusion appears to be one of the earliest events involving prostate cancer invasion and leads to the over expression of the fused ETS gene in an androgen-regulated manner. The finding have now been confirmed by other groups.21, 22 There is still much to be learned about this common prostate cancer gene fusion. Although we have recently described the common intronic deletion on chromosome 21 associated with the TMPRSS2:ERG gene fusion,23 The DNA breakpoint(s) have not yet been identified but would help in the development of diagnostic tools for prostate cancer. The exact frequency of the TMPRSS2:ETS fusion still needs to be determined in population-based studies. The high percentage of TMPRSS2:ERG fusion prostate cancers suggests that ERG may be the most common fusion partner. The hospital-based studies to date suggest that at least 50% of prostate cancers harbor the TMPRSS2:ERG gene fusion. With the recent identification of a third molecular subtype (TMPRSS2:ETV4),20 one can anticipate finding other translocation partners such as FLI1 based on expression array data. This would be similar to observation in the Ewing's family tumors, where approximately 85% of tumors harbor a tumor-associated t(11;22)(q24;q12) rearrangement resulting in the juxtaposition of the EWS gene (Ewing's Sarcoma Gene) on chromosome 22 with the FLI1 gene on chromosome 11. Four other ETS family members have been identified as translocation partners of EWS. The second most common ETS translocation partner is ERG seen in approximately 10% of cases.24 Finally, the identification of the TMPRSS2:ETS gene fusion in prostate cancer suggests that distinct molecular subtypes may further define risk for disease progression.
The discovery of the common TMPRSS2:ETS gene fusions in prostate cancer using COPA suggest that other translocations may be identified in common epithelial tumors. The combination of an organ specific promoter such as TMPRSS2 for prostate cancer fused to an oncogene may also be a common theme in carcinogenesis. COPA has now begun a new search for targetable fusion products. Perhaps, leading to rational drug development similar to the development of imatinib (STI571, Gleevec) therapy for CML.
References
Luo J, Duggan DJ, Chen Y, et al. Human prostate cancer and benign prostatic hyperplasia: molecular dissection by gene expression profiling. Cancer Res 2001;61:4683–4688.
Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature 2001;412:822–826.
Luo J, Dunn T, Ewing C, et al. Gene expression signature of benign prostatic hyperplasia revealed by cDNA microarray analysis. Prostate 2002;51:189–200.
Rhodes DR, Barrette TR, Rubin MA, et al. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res 2002;62:4427–4433.
Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004;6:1–6.
Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644–648.
Bittner M, Meltzer P, Chen Y, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000;406:536–540.
Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 2004;350:1617–1628.
Vasselli JR, Shih JH, Iyengar SR, et al. Predicting survival in patients with metastatic kidney cancer by gene-expression profiling in the primary tumor. Proc Natl Acad Sci USA 2003;100:6958–6963.
Segal NH, Pavlidis P, Noble WS, et al. Classification of clear-cell sarcoma as a subtype of melanoma by genomic profiling. J Clin Oncol 2003;21:1775–1781.
Ross ME, Zhou X, Song G, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 2003;102:2951–2959.
Lapointe J, Li C, Higgins JP, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA 2004;101:811–816.
Tian E, Zhan F, Walker R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 2003;349:2483–2494.
Dhanasekaran SM, Dash A, Yu J, et al. Molecular profiling of human prostate tissues: insights into gene expression patterns of prostate development during puberty. FASEB J 2005;19:243–245.
Wang Y, Klijn JG, Zhang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005;365:671–679.
Welsh JB, Sapinoso LM, Su AI, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res 2001;61:5974–5978.
Zhan F, Hardin J, Kordsmeier B, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002;99:1745–1757.
Cheok MH, Yang W, Pui CH, et al. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat Genet 2003;34:85–90.
Wigle DA, Jurisica I, Radulovich N, et al. Molecular profiling of non-small cell lung cancer and correlation with disease-free survival. Cancer Res 2002;62:3005–3008.
Tomlins SA, Mehra R, Rhodes DR, et al. TMPRSS2:ETV4 gene FUSIONS define a third molecular subtype of prostate cancer. Cancer Res 2006;66:3396–3400.
Soller MJ, Isaksson M, Elfving P, et al. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes Chromosomes Cancer 2006;45:717–719.
Yoshimoto M, Joshua AM, Chilton-Macneill S, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia 2006;8:465–469.
Perner S, Demichelis F, Beroukhim R, et al. TMPRSS2:ERG fusion associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res 2006;66:8337–8341.
Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294–299.
Acknowledgements
We thank Scott A Tomlins, Daniel Rhodes, and Francesca Demichelis for their help in developing this review.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Rubin, M., Chinnaiyan, A. Bioinformatics approach leads to the discovery of the TMPRSS2:ETS gene fusion in prostate cancer. Lab Invest 86, 1099–1102 (2006). https://doi.org/10.1038/labinvest.3700477
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700477
Keywords
This article is cited by
-
The oncogene ERG: a key factor in prostate cancer
Oncogene (2016)
-
Maintaining and reprogramming genomic androgen receptor activity in prostate cancer
Nature Reviews Cancer (2014)
-
Mapping of TMPRSS2–ERG fusions in the context of multi-focal prostate cancer
Modern Pathology (2008)
