
Abstract
Heme oxygenase-1 (HO-1) is the inducible isoform of an enzyme family responsible for heme degradation and was suggested to be involved in the acute phase response in the liver. However, the mechanisms of the HO-1 regulation under inflammatory conditions are poorly understood. Therefore, the purpose of the current work was to study the expression of HO-1 in the liver and other organs of rats with a localized inflammation after intramuscular injection of turpentine oil (TO). Since interleukin-6 (IL-6) is known to be a principal mediator of inflammation, the levels of this cytokine were also estimated in the animal model used. HO-1 and IL-6 expression was evaluated by Northern blot, in situ hybridization, Western blot, immunohistochemistry and enzyme-linked immunosorbent assay. In the liver and injured muscle, the HO-1 mRNA levels were dramatically increased 4–6 h after TO administration. HO-1 protein levels in the liver were elevated starting from 6–12 h after the treatment. In other internal organs such as the heart, kidney and large intestine, only a slight induction of HO-1 mRNA was observed. IL-6-specific transcripts appeared only in the injured muscle and were in accordance with serum levels of IL-6. In turn, temporal expression of IL-6 in the muscle and circulatory IL-6 levels correlated well with HO-1 expression in the liver and injured muscle. In the liver of control rats HO-1 protein was detected in Kupffer cells, while in TO-injected rats also hepatocytes became strongly HO-1 positive. Conversely, in the injured muscle, HO-1 immunoreactivity was attributed only to macrophages. Our data demonstrate that during localized inflammation HO-1 expression was rapidly and strongly induced in macrophages of injured muscle and in hepatocytes, and IL-6 derived from injured muscle seems to be responsible for the HO-1 induction in the liver.
Similar content being viewed by others


Main
Heme oxygenase (HO, EC 1.14.99.3) is the microsomal enzyme that catalyses the initial reaction in heme catabolism yielding equimolar amounts of biliverdin, carbon monoxide, and free divalent iron.1, 2 Biliverdin is subsequently reduced to bilirubin by biliverdin reductase, while the free iron is promptly sequestered into ferritin. Three isoforms of HO (HO-1, HO-2 and HO-3) have been described in mammals as the products of separate genes.3, 4, 5 HO-1 is widely expressed in tissues and highly inducible in virtually all cell types by a variety of stimuli.6 Constitutively expressed HO-2 is unresponsive to any of the known HO-1 inducers.5, 6, 7 The recently discovered HO-3 isozyme is nearly devoid of catalytic activity and serves mainly as heme-sensing/binding protein.4 HO-1 expression is induced not only in response to its substrate heme,8 but also by various stress stimuli such as hydrogen peroxide, xenobiotics, nitric oxide, ultraviolet-A radiation, heavy metals, hypoxia, hyperoxia, heat shock,9, 10 endotoxin11, 12 and proinflammatory cytokines.13, 14, 15, 16 Accumulating evidence reveals the protective role of HO-1 by virtue of the antiinflammatory, antiapoptotic and antiproliferative activities of the products released by the heme breakdown reaction.17, 18, 19
The acute phase response (APR) is a defense reaction of the organism directed to restrict the area of damage and to eliminate, or at least isolate, the damaging agent. Every agent that leads to loss of tissue integrity induces a local reaction known as inflammation.20 Cells of the inflammatory infiltrate produce and/or induce the production of cytokines, the main soluble factors responsible for the onset, progression and resolution of the APR, whereas the liver is the primary target organ of the host defense reaction.21 The liver parenchymal cells, hepatocytes, are the source of specific circulatory proteins, called acute phase proteins (APP), for example, α2-macroglobulin, C-reactive protein, serum amyloid A, etc. The plasma concentrations of APPs are dramatically changed during the APR.20 Interleukin-6 (IL-6), interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) are the most potent inducers of APP synthesis in hepatocytes,20 and are thereby considered to be the main mediators of the APR.
Although it has been shown that HO-1 could be upregulated under inflammatory conditions in the liver15, 16, 22 and in hepatoma cell lines,14 it remains unclear whether induction of HO-1 in vivo occurs as a response to cytokines released locally at the site of inflammation. To test this hypothesis, we analyzed HO-1 expression in the liver and extrahepatic tissues of rats after induction of sterile muscle abscess by injection of turpentine oil (TO), which subsequently leads to a local inflammation followed by systemic APR.23
Materials and methods
Animals
Male Wistar rats (about 200 g body weight) were purchased from Harlan–Winkelmann (Borchen, Germany) and kept under standard conditions with 12-h light/dark cycles and access to fresh water and food pellets ad libitum. All animals received humane care in accordance with the institution's guidelines, the German Convention for Protection of Animals and the National Institutes of Health guidelines.
Materials
All chemicals were of analytical grade and obtained from commercial sources as indicated: [α-32P]-labeled deoxy-cytidine-triphosphate (specific activity 3000 Ci/mmol) and Hybond N nylon membranes were from Amersham Pharmacia Biotech (Freiburg, Germany); Protran nitrocellulose membranes from Schleicher and Schuell (Dassel, Germany); pentobarbital sodium (Narcoren) from Merial (Hallbergmoos, Germany). TO was purchased from the University Pharmacy Goettingen (Goettingen, Germany). Media M199, collagenase type I and fetal calf serum were from Biochrom (Berlin, Germany); T3 and T7 RNA polymerases, DNase I, RNase A and yeast tRNA from Roche (Mannheim, Germany). Nycodenz was obtained from Nyegaard (Oslo, Norway), hybridization solution QuikHyb from Stratagene (Heidelberg, Germany), agarose, guanidine isothiocyanate and Taq DNA-polymerase were from Invitrogen (Karlsruhe, Germany). L-glutamine was from PAA (Linz, Austria); cesium chloride from Paesel and Lorei (Frankfurt, Germany). Lipopolysaccharide (LPS) from Salmonella minnesota was purchased from Difco Laboratories (Detroit, MI, USA). All other reagents and chemicals were from Sigma-Aldrich Chemie (Munich, Germany) or Merck (Darmstadt, Germany).
cDNA Probes
Two cDNA probes used for Northern blot analysis were amplified from respective clones by polymerase chain reaction using standard T3 and T7 primers: an 883 base pair (bp) EcoRI-HindIII insert of pBS-HO-124 and a 901 bp PstI-BamHI fragment of a rat IL-6 clone.25 In order to control APR in rat liver, a rat α2-macroglobulin cDNA clone26 and a clone carrying a 700 bp fragment of mouse albumin cDNA27 were used. To validate quantitative Northern blot results, a clone carrying the rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA,28 or an oligonucleotide (5′ AAC GAT CAG AGT AGT GGT ATT TCA CC 3′) complementary to 28S rRNA,29 was used.
Antibodies
Rabbit polyclonal antiserum against rat HO-1 and mouse anti-rat HO-1 monoclonal antibody were purchased from Stressgen Biotechnologies Corp. (Victoria, BC, Canada). Mouse anti-rat ED1 and ED2 monoclonal antibodies were from Serotec (Dusseldorf, Germany) and mouse monoclonal antibody against β-actin was obtained from Sigma-Aldrich Chemie. Horseradish peroxidase-conjugated donkey anti-rabbit and rabbit anti-mouse immunoglobulins from Amersham Pharmacia Biotech and from Dako (Copenhagen, Denmark), respectively, were used as secondary antibodies.
Isolation and Culture of Rat Kupffer Cells
Rat liver macrophages (Kupffer cells) were isolated according to the method of Knook and Sleyster30 as previously described.31 After assessment of viability by trypan blue staining, cells were taken up in culture medium (M199, 15% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1% L-glutamine) and plated onto 60-mm Falcon plastic dishes at a density of 6 × 106 cells per dish. Cell cultures were maintained at 37°C in a 95% air/ 5% CO2 atmosphere and saturated humidity. Treatment of Kupffer cells with LPS (500 ng/ml) was carried out in serum-reduced medium (0.3% fetal calf serum) for 24 h. Afterwards, the cells were washed with phosphate-buffered saline (PBS), pH 7.4 and frozen at −80°C for subsequent RNA isolation. Total cellular RNA isolated from LPS-treated Kupffer cells served as a positive control for detection of IL-6-specific transcripts.
Induction of Acute Phase Reaction
Acute phase reaction was induced by intramuscular injection of 5 ml/kg TO into the right and left hind limb of ether-anesthetized rats; control animals received no injection. Animals were killed at time points ranging from 30 min to 48 h after TO administration under pentobarbital anesthesia. Different organs as well as hind limb muscle tissue including the TO-injected area were excised, rinsed with physiological sodium saline, frozen in liquid nitrogen and stored at −80°C until use.
Preparation of Tissue Homogenates
About 100 mg of frozen tissue was homogenized with Ultra-Turrax TP 18/10 three times for 10 s each in 10 volumes of 50 mM Tris-HCl buffer, pH 7.4, containing 150 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid, 1% Triton X-100, 1 mM phenylmethanesulfonyl fluoride, 1 mM benzamidine, 1 μg/ml leupeptin, 10 μM chymostatin, 1 μg/ml antipain, 1 μg/ml pepstatin A. The entire procedure was carried out at 4°C. Crude homogenates were passed five times through a 22 G needle applied to a syringe, centrifuged at 4°C for 5 min at 10 000 × g and the protein concentration was determined in supernatants using the BCA protein assay reagent kit (Pierce, Bonn, Germany). Aliquots of the homogenates were stored at −20°C until use.
Western Blot Analysis
Protein samples were denatured in electrophoresis buffer (50 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate, 10% glycerol, 50 μg/ml bromphenol blue, 2% β-mercaptoethanol) at 95°C for 10 min and 15 μg of total protein was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis with a 12.5% resolving gel according to the method of Laemmli32 using a Mini-PROTEAN™ III Electrophoresis Cell (BioRad, Munich, Germany). As molecular weight standards, the Rainbow™ colored protein markers (Amersham Pharmacia Biotech) were used. Proteins were transferred onto nitrocellulose membranes in a Mini-Trans-Blot® Transfer Cell (BioRad) according to the procedure of Towbin et al.33 The membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) and 5% nonfat dry milk at 4°C overnight and processed for immunodetection using a rabbit polyclonal antiserum against HO-1 (1:2000 dilution in TBS-T with 0.5% milk) for 2 h at room temperature. After a subsequent washing step, peroxidase-conjugated anti-rabbit immunoglobulin was used as a secondary antibody. Visualization of immunoreactive bands was performed using the SuperSignal® West Pico Chemiluminescent Substrate (Pierce) and the signal was detected by short exposure to X-ray film (Fuji, Duesseldorf, Germany). To ensure equal loading of proteins, the blots were stripped and reprobed with anti-β-actin antibody (1:5000 dilution in TBS-T with 0.5% milk; 1 h, room temperature).
Immunohistochemistry
For immunohistochemistry, tissue specimens from rat liver and muscle were rinsed with 0.9% NaCl and snap-frozen in liquid nitrogen. Cryostat sections (5 μm) were air-dried, fixed with methanol (−20°C, 10 min) and acetone (−20°C, 10 s), and stored at −20°C. After inhibition of endogenous peroxidase by incubating the slides with PBS containing glucose/glucoseoxidase/sodium azide, they were treated with FCS for 30 min to minimize nonspecific staining. The sections were incubated in a humidified chamber with the first antibody directed against HO-1, ED1 and ED2 diluted in PBS at the ratios of 1:500, 1:100, and 1:100, respectively, for 1 h at room temperature. Negative controls were incubated with normal mouse serum instead of the first antibody. After washing, the slides were covered with peroxidase-conjugated anti-mouse immunoglobulin preabsorbed with normal rat serum to avoid crossreactivity. Slides were washed and incubated with PBS containing 3,3’-diaminobenzidine (0.5 mg/ml) and H2O2 (0.01%) for 10 min to visualize immune complexes. Nuclei were counterstained with Meyer's hemalum solution before the slides were mounted with coverslips.
Enzyme-Linked Immunosorbent Assay (ELISA)
Blood samples from the inferior vena cava were collected from control and treated rats, allowed to clot overnight at 4°C and centrifuged for 20 min at 2000 × g. Serum was removed and stored in aliquots at −20°C. Serum IL-6 concentrations were determined by rat IL-6-specific ELISA, standardized with rat IL-6 using the Quantikine® M immunoassay kit (R&D Systems, Wiesbaden, Germany) according to the manufacturer's instructions.
RNA Extraction and Northern Blot Analysis
Total RNA was isolated from different rat tissues including the liver, spleen, kidney, lung, heart, skeletal muscle, small and large intestine by means of guanidine isothiocyanate extraction, cesium chloride density-gradient ultracentrifugation and ethanol precipitation according to method of Chirgwin et al.34 The RNA obtained was quantified by measuring the absorbance at 260 nm. Total RNA (5–10 μg/lane) was size-fractionated by electrophoresis in 1% agarose–formaldehyde gels, transferred to nylon membranes using the capillary transfer system and crosslinked by ultraviolet light.
Each cDNA probe was labeled with 40–60 μCi of [α-32P]-labeled deoxy-cytidine-triphosphate (3000 Ci/mmol) by nick translation while the 28S rRNA oligonucleotide was labeled by random priming using commercially available kits according to the manufacturers’ instructions. The probes were purified from unincorporated radiolabeled nucleotides on NICK™ Columns prepacked with Sephadex® G-50 (Amersham Pharmacia Biotech).
The membranes were hybridized with respective [32P]-labeled cDNA probes (1 × 106 cpm/ml) in QuikHyb hybridization solution at 68°C for 1 h. After hybridization the membranes were washed in 2 × standard saline citrate (SSC) containing 0.1% sodium dodecyl sulfate twice for 15 min at room temperature and twice for 5–15 min at 50°C. Nylon filters were then exposed to X-Omat AR films (Kodak, Rochester, NY, USA) with an intensifying screen for 1–3 days at −80°C. The autoradiograms were scanned (Bio-Rad, Hercules, CA, USA) and densitometrically analyzed (Molecular Analyst, Bio-Rad). To ensure equal loading of RNA, the membranes were stripped and reprobed for 28S rRNA or GAPDH mRNA.
In Situ Hybridization
In situ hybridization (ISH) was performed according to the published protocol35 with slight modifications. Briefly, antisense and sense riboprobes were synthesized with the DIG RNA labeling kit (Roche) using T3 and T7 polymerases, respectively, and a plasmid containing rat IL-6 cDNA25 as a template. Cryostat sections (5 μm) were immediatelly fixed with 4% paraformaldehyde diluted in PBS. To inactivate endogenous RNases, slides were treated with 0.1% diethyl pyrocarbonate/PBS solution. Sections were prehybridized with hybridization solution (50% formaldehyde, 1 mM dithiothreitol, 5 × SSC, 500 ng/ml yeast tRNA, 10% dextran sulfate) for 2 h at 50°C followed by overnight hybridization with the hybridization solution containing labeled antisense or sense riboprobe (500 ng/ml) at 50°C. After sequential washing steps with 2 × SSC and 0.1 × SSC for 15 min at 50°C, the slides were treated for 30 min at 37°C with RNase A (10 μg/ml) diluted in 2 × SSC followed by an additional washing step with 0.1 × SSC for 15 min at 50°C. Signal detection was carried out by the DIG nucleic acid detection kit (Roche) according to the manufacturer's instructions. Nuclei were counterstained with methyl green solution before the slides were mounted with coverslips.
Statistical Analysis
Data are presented as means±standard error of the mean (s.e.m.). A P-value of less than 0.05 (Student's t-test) was considered significant. Only statistically significant values of differences are indicated.
Results
HO-1 Distribution in the Livers of Untreated and TO-Treated Rats
To ensure that the intramuscular TO injection used for the APR induction did not cause liver damage, hematoxylin/eosin staining was performed. The staining did not reveal any morphological difference between the livers of control and TO-treated rats (data not shown).
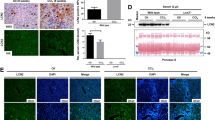
Cell type-specific expression of HO-1 in the livers obtained from control and TO-treated rats was assessed by immunohistochemical analysis of sequential cryostat sections. Under physiological conditions, the expression of HO-1 immunoreactive protein was mainly attributed to Kupffer cells, as verified by the application of an antibody against the rat ED2, a well-known specific marker of resident liver macrophages (Kupffer cells), whereas liver parenchymal cells were HO-1 negative (Figure 1a, c, d). However, during the TO-induced inflammatory response, a prominent induction of HO-1 immunoreactivity was observed in the liver parenchyma, whereas no significant increase in the intensity and number of the HO-1-positive Kupffer cells was visible (Figure 1b). The negative controls demonstrated specificity of the immunohistological analysis performed (Figure 1e, f).

Immunohistochemical analysis of the livers from control and TO-treated rats. Sequential cryostat sections (5 μm) of control rat liver (a, c, e) or liver excised from the rat 12 h after TO administration (b, d, f) were stained with monoclonal antibodies against rat HO-1 (a, b) or ED2 (c, d) as described in Materials and methods. Horseradish peroxidase-linked anti-mouse immunoglobulin was applied as a secondary antibody. The positive cells are brown colored. Nuclei are counterstained in blue. The panels e and f represent negative controls obtained by substitution of primary antibody with normal mouse serum, black arrows indicate central veins, white arrow—periportal field. Original magnification × 100.
These data suggest that under control conditions hepatic HO-1 is mainly expressed in Kupffer cells, whereas during TO-induced APR HO-1 expression is also induced in hepatocytes.
HO-1 Distribution in Injured Muscle during TO-Induced Acute Phase Reaction
Cell type-specific expression of HO-1 in normal and injured muscle from TO-treated rats was assessed by immunohistochemical analysis of sequential cryostat sections. Normal muscular tissue did not demonstrate any HO-1 positivity (Figure 2a). However, under inflammatory conditions HO-1 immunoreactive protein appeared in some small, elongated, irregularly shaped cells infiltrating injured muscle, whereas muscle fibers were HO-1 negative (Figure 2b). Interestingly, the muscle tissue adjacent to the necrotic areas remained intact and did not show any positive reaction with anti-HO-1 antibody (data not shown).

Immunohistochemical analysis and in situ hybridization of normal and injured muscular tissue from rat hind limb. Sequential cryostat sections (5 μm) of normal (a, c) or injured (6 h after TO administration) rat muscle (b, d) were stained with monoclonal antibodies against rat HO-1 (a, b) or ED1 (c, d) as described in Materials and methods. Horseradish peroxidase-linked anti-mouse immunoglobulin was applied as a secondary antibody. The positive cells are brown colored. Nuclei are counterstained in blue. Original magnification × 250. After paraformaldehyde fixation and inactivation of endogenous RNases, cryostat sections of normal (e) or injured (6 h after TO administration) rat muscle (f) were subjected to in situ hybridization with digoxigenin-labeled IL-6-specific antisense riboprobe as described in Materials and methods. Hybridization with the sense riboprobe was performed in parallel and served as negative control (data not shown). The positive cells are dark-blue colored. Nuclei are counterstained in light-green. Black arrows indicate the cells infiltrating the injured muscle and demonstrating strong positivity for IL-6-specific transcripts. Original magnification × 250.
Application of an antibody against the macrophage-specific marker ED1 using respective subsequent sections (Figure 2d) demonstrated colocalization of HO-1 and ED1. Notably, no ED1 positive cells were observed in normal muscular tissue (Figure 2c). These data suggest that HO-1 expression is most likely attributed to macrophages recruited to muscular tissue during inflammation.
Expression of IL-6 mRNA Occurred Only in Muscle during TO-Induced Acute Phase Reaction
IL-6 is known as a key cytokine released during TO-induced APR in the rat.36 In order to determine the source of IL-6 under the experimental conditions, Northern blot analysis of total RNA prepared from skeletal muscle, liver, lung, heart, kidney, spleen, small and large intestine, at various time points after injection of TO, was performed. Total cellular RNA (10 μg) isolated from rat Kupffer cells treated with LPS (500 ng/ml) was processed in parallel and served as a positive control for IL-6-specific transcripts.37 The 1.2 and 2.4 kb IL-6-specific transcripts were detectable only in the injured skeletal muscle from 2 to 12 h after TO injection with maximal levels between 4 and 6 h (Figure 3). In accordance with these results, ISH data demonstrate that in the injured muscle, inflammatory cells infiltrating muscular tissue were the major source of IL-6 transcripts (Figure 2f). In contrast, ISH did not reveal any significant IL-6 mRNA expression in skeletal muscle obtained from control animals (Figure 2e) as well as in the muscle tissue adjacent to the TO-injected areas (data not shown). Other rat tissues were IL-6 negative as demonstrated by Northern blot analysis suggesting that only injured muscle is the source of IL-6 during TO-induced APR.

Time course of IL-6 mRNA expression in various rat tissues after TO administration. APR in the rat was induced by TO injection, and total RNA from skeletal muscle, liver, lung, heart, kidney, spleen, small and large intestine was extracted at the indicated time points as described in Materials and methods. In all, 10 μg of total RNA from these tissues was subjected to Northern blot analysis and hybridized with [32P]-labeled cDNA specific for IL-6. An oligonucleotide complementary to 28S rRNA was used to ensure equal loading of RNA (data not shown). Total cellular RNA (10 μg) isolated from rat Kupffer cells treated with LPS (500 ng/ml) was processed in parallel and served as a positive control for IL-6-specific transcripts.
Serum IL-6 Levels in Rat during TO-Induced Acute Phase Reaction
To elucidate the possible relationship between hepatic HO-1 induction and IL-6 mRNA expression in the injured muscle, circulating IL-6 levels in TO-treated rats were measured by rat IL-6-specific ELISA. Serum IL-6 concentrations were markedly elevated during TO-induced APR with maximal values 1725.785±310.985 and 1277.913±383.983pg/ml at 6 and 12 h after injection, respectively. This corresponds to about 115- and 85-fold increase compared with control levels (Figure 4). It should be noted that serum IL-6 levels were already elevated 2 h after TO administration with an about six-fold increase compared with the controls. Serum IL-6 levels in TO-treated rats correlated well with IL-6 gene expression in the injured muscle confirming that inflamed muscle seems to be the only source of IL-6 in the model under study.

Enhanced serum IL-6 levels in rats after TO administration. APR in the rat was induced by TO injection, and blood was collected at the indicated time points after injection as described in Materials and methods. Serum IL-6 concentrations were measured by ELISA. The results are presented as mean±s.e.m. (n=3).
Expression of Hepatic HO-1 mRNA and Protein during TO-Induced Acute Phase Reaction
Liver specimens were collected from the control and treated animals at the time points ranging from 0.5 to 48 h and subjected to RNA and protein extraction. As revealed by Northern blot analysis, HO-1 mRNA levels were substantially increased 4–6 h after TO administration with a maximum at 6 h (Figure 5a). Since α2-macroglobulin is a well-known positive, and albumin α negative, acute phase protein in rats,38 hybridizations with α2-macroglobulin- and albumin-specific cDNAs were performed to ensure the APR induction in the treated animals. The α2-macroglobulin mRNA levels were elevated from 4 to 24 h and albumin mRNA decreased reaching lowest levels 24–36 h after TO injection (Figure 5a), which is in accordance with published data.39

Time course of hepatic HO-1 mRNA and protein expression after TO administration. APR in the rat was induced by TO injection, liver specimens were subsequently removed at the indicated time points and homogenized for protein and RNA extraction as described in Materials and methods. (a) Total RNA (10 μg) from the livers of control and TO-treated rats was subjected to Northern blot analysis and sequentially hybridized with [32P]-labeled cDNAs specific for HO-1, α2-macroglobulin (α2-M), albumin and GAPDH (used as an internal loading control). The sizes of the transcripts (in kilobases, kb) are indicated on the left. A representative of four independent experiments is shown. (b) Proteins from the livers of control and TO-treated rats were size-fractionated by electrophoresis, transferred to nitrocellulose membrane and probed with a rabbit polyclonal antiserum against HO-1 as described in Materials and methods. Mouse monoclonal antibody against β-actin was used to ensure equal loading of proteins.
The induction of HO-1 gene expression was followed by the enhancement of HO-1 protein synthesis as shown by Western blot analysis. HO-1 protein levels started to increase after 4 h and remained elevated up to 48 h after TO administration (Figure 5b) indicating that in rat liver the lifetime of the HO-1 protein under inflammatory conditions seems to be considerably longer than that of the HO-1 mRNA.
Expression of HO-1 mRNA in Extrahepatic Rat Tissues during TO-Induced Acute Phase Reaction
To determine whether HO-1 induction is a liver-specific response to localized inflammation or it could also occur in other rat tissues under the given experimental conditions, Northern blot analysis of total RNA obtained from skeletal muscle, lung, heart, kidney, spleen, small and large intestine at various time points after injection of TO was performed using radiolabeled HO-1 cDNA as a probe. A strong induction of HO-1 mRNA was observed in the skeletal muscle from the site of injection where the APR was initiated by local inflammation. The HO-1-specific transcripts appeared already at 2 h, reached maximal levels by 4–6 h after TO injection and then slowly decreased (Figure 6). We have also found out that in the heart, kidney and large intestine, slight induction of HO-1 mRNA was observed 6 h after TO injection. Notably, the spleen showed relatively high basal levels of HO-1 gene expression, which is most likely attributed to resident macrophages highly abundant in this organ (Figure 6).

Time course of HO-1 mRNA expression in extrahepatic rat tissues after TO administration. APR in the rat was induced by TO injection, and total RNA from skeletal muscle, lung, heart, kidney, spleen, small and large intestine was extracted at the indicated time points as described in Materials and methods. Total RNA (10 μg) from these tissues was subjected to Northern blot analysis and hybridized with [32P]-labeled cDNA specific for HO-1. An oligonucleotide complementary to 28S rRNA was used to ensure equal loading of RNA (data not shown).
Discussion
The present study has demonstrated that during the acute phase reaction induced by TO injection in rats, HO-1 expression is substantially upregulated only in the liver, the organ mostly responsible for the APR, and in the injured muscle, the site of localized inflammation. The significant induction of HO-1 in the liver was attributed to hepatocytes, whereas macrophages represented the major source of HO-1 in the injured muscle. Under inflammatory conditions, muscular tissue at the site of TO injection was the only source of IL-6, the principal mediator of the APR in rats. The upregulation of IL-6 mRNA expression was accompanied by elevated serum IL-6 levels and HO-1 induction in the liver suggesting that during TO-induced APR, IL-6 derived from the injured muscle is responsible for the HO-1 induction in hepatocytes.
Regulation of HO-1 Expression in TO Model of the APR in Rats
In the current study, TO injection in rats was used as a model system to investigate HO-1 regulation during the APR. The results obtained demonstrate that HO-1 mRNA expression was dramatically increased in the liver by 4–6 h after TO administration with a peak at 6 h (Figure 5a). Previously, hepatic HO-1 mRNA expression in a TO model was studied by Lyoumi and colleagues22, 40 along with an LPS-induced model of the APR. Using reverse transcription-polymerase chain reaction, they also have shown that in the liver HO-1-specific transcripts were maximally elevated 6 h after TO administration; no information, however, was provided about the protein. In the present study, we did not only investigate HO-1 mRNA expression but also HO-1 protein levels, and we have demonstrated that HO-1 protein was markedly upregulated after 6 h and reached maximal levels 24 h after TO injection (Figure 5b).
To find out whether the induction of HO-1 during inflammation is a liver-specific direct consequence of the APR, in the current study, in addition to the liver, HO-1 expression in extrahepatic sites during TO-induced APR was investigated for the first time. Besides the liver, which is the organ mostly responsible for the APR, the most prominent HO-1 mRNA induction occurred in skeletal muscle, where the APR was initiated by TO injection (Figure 6). In the other internal organs investigated, only moderate changes in HO-1 expression were observed (Figure 6).
It should be noted that despite the similarities in general signs and symptoms, different in vivo models of the APR are characterized by divergent patterns of mediators and plasma proteins released during inflammation.20, 41 For instance, an LPS model is characterized by rapid circulatory increase of major proinflammatory cytokines (IL-6, IL-1β and TNF-α) leading to systemic inflammation.42, 43, 44 The variety of mediators in this model makes it difficult to define the role of individual cytokines in the induction of HO-1 in the liver. In this regard, the TO-induced model of the APR is of particular interest because it is accompanied by an increase of circulating IL-6 levels, whereas plasma IL-1β and TNF-α levels remain undetectable.36
Expression of IL-6 after TO Administration and Its Possible Role in HO-1 Induction in the Liver
Since IL-6 is the principal mediator of the APR in the liver,45 we hypothesized that also TO-dependent hepatic HO-1 induction was mediated mainly by IL-6. An increased secretion of IL-6 into the bloodstream in TO model has been reported before.36 Furthermore, it has also been shown that IL-6-deficient mice were unable to maintain a normal inflammatory response to localized tissue damage generated by TO injection.41 In agreement with these findings, we have demonstrated that serum IL-6 concentrations were markedly elevated after TO injection (Figure 4).
To find the source of IL-6 released during TO-induced APR, we studied IL-6 mRNA expression in various rat tissues. Our data have clearly shown that during TO-induced APR, injured skeletal muscle was the only organ where induction of IL-6 expression was observed (Figure 3), and as demonstrated by ISH, inflammatory cells appear to be the major source of IL-6 in the injured muscle (Figure 2f). Furthermore, temporal expression of IL-6 in the muscle correlated well with circulatory IL-6 levels. Remarkably, IL-6-specific transcripts appeared in the skeletal muscle 2 h after TO injection, whereas induction of HO-1 mRNA expression in the liver was observed 4–6 h after TO administration. Moreover, our preliminary in vitro experiments demonstrated significant induction of HO-1 mRNA and protein levels in primary cultured rat hepatocytes after IL-6 treatment (data not shown). Taken together, these data suggest that induction of HO-1 expression in the liver during the APR is a consequence of the induced IL-6 expression at the site of inflammation followed by the increased serum IL-6 levels.
Interestingly, the serum IL-6 levels were still high 12 h after TO injection (Figure 4), while HO-1 mRNA levels in the liver were already decreased (Figure 5a). However, it is well known that IL-6 induces downregulation of its cognate receptor, IL-6R, in vitro.46 Moreover, it has also been demonstrated that IL-6R mRNA expression in rat liver markedly decreased by 6–12 h after TO administration.47 Therefore, it is likely that the dramatic increase of IL-6 levels observed in the TO-induced APR model subsequently leads to downregulation of the IL-6R, thus abolishing the action of IL-6 in the liver by a negative feedback mechanism.
Cell-Type Specificity of HO-1 Induction and Its Possible Role in TO-Induced APR
To identify cell types responsible for the TO-induced HO-1 expression in the injured muscle and in the liver, immunohistochemical analysis was performed. It has been demonstrated that within the injured skeletal muscle, synthesis of HO-1 occurred mainly in the cells positive for ED1, a known marker of monocytes and macrophages, whereas muscle fibers were HO-1 negative (Figure 2b, d). These findings are consistent with the data obtained by Kampfer and colleagues, who have demonstrated that infiltrated macrophages are the major source of HO-1 at the wound site in the mouse excisional model of skin repair. The authors assumed involvement of HO-1 in the regulation of macrophage-derived cytokine release.48 Indeed, numerous recent observations point to a potential role of HO-1 in the downregulation of proinflammatory cytokine expression at the site of inflammation in various model systems.49, 50, 51, 52, 53 It has also been shown that HO-1 might provide an anti-inflammatory action in vivo through the downmodulation of cell adhesion molecules accompanied by inhibition of leukocyte infiltration,54, 55, 56 and via promotion of noninflammatory angiogenesis facilitating tissue repair.56 Moreover, an increased leukocyte adhesion to the vascular wall and spontaneous perivascular infiltration of leukocytes in various tissues of HO-1-deficient mice has been reported before.57
Although hepatic HO-1 production under physiological, and some pathological, conditions is ascribed mainly to Kupffer cells, the resident liver tissue macrophages,58, 59, 60, 61 there is evidence that HO-1 can also be upregulated under certain conditions in hepatocytes as it has been shown in vivo by several groups.58, 62, 63, 64 Immunohistochemical analysis of normal rat liver carried out in the present study revealed that expression of HO-1 was attributed predominantly to Kupffer cells (Figure 1a, c), which is in agreement with the observations of other groups.58, 59 However, the induction of HO-1 immunoreactivity in the liver parenchyma after TO administration has been shown for the first time (Figure 1b). This suggests that during the inflammatory response to localized tissue injury, hepatocytes are a significant source of increased HO-1 mRNA and protein expression levels observed in the livers of TO-treated rats (Figure 5).
The data regarding muscle- and liver-specific HO-1 induction in the TO model of the APR might be of physiological importance, since the products of HO-1 reaction have been shown to be involved in a wide range of cytoprotective and anti-inflammatory mechanisms. Carbon monoxide (CO) regulates blood vessel tone,65 prevents platelet activation and aggregation,66 suppresses production of proinflammatory cytokines and contributes to interleukin-10 upregulation in macrophages both in vitro and in vivo.67 Moreover, CO mediates the anti-inflammatory effects of interleukin-10.68 It has also been reported that CO prevents apoptosis in several cell types, including endothelial cells, fibroblasts and hepatocytes.18 Biliverdin and bilirubin are potent antioxidants and may protect cells from oxidative injury by scavenging reactive oxygen species.69 Biliverdin has also been shown to inhibit human complement system in vitro.70 Bilirubin had a protective effect on the transplanted liver grafts via inhibition of lipid peroxidation in hepatocytes.71 Although reduced iron (Fe2+) released by HO-1 from the core of the heme molecule is a potent pro-oxidant, the potential catalysis of oxidative reactions by this compound is limited through its chelation by iron-sequestering protein ferritin as well as via its active removal from the cell by the specific Fe-ATPase pump.72 Moreover, ferritin production and biological activity of Fe-ATPase pump are potentiated by increased free iron in the intracellular pool due to enhanced HO-1 expression.73, 74
In summary, our data demonstrate that in rats, during a localized inflammation, the expression of HO-1 is rapidly and strongly induced in macrophages of the injured muscle and in liver parenchyma. IL-6 derived from inflammatory cells at the injured muscle seems to be responsible for the HO-1 induction in the liver but not in the other internal organs during the TO-induced APR. In this regard, HO-1 might be referred to as an early intracellular acute phase protein. Taken together, these data point to an important role of HO-1 in cytoprotection of hepatocytes, which might become damaged during their intensive function under inflammatory conditions. Furthermore, at the site of local injury, upregulation of HO-1 in macrophages can participate in the resolution of inflammation.
References
Tenhunen R, Marver HS, Schmid R . The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 1968;61:748–755.
Tenhunen R, Marver HS, Schmid R . Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem 1969;244:6388–6394.
Maines MD, Trakshel GM, Kutty RK . Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 1986;261:411–419.
McCoubrey Jr WK, Huang TJ, Maines MD . Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem 1997;247:725–732.
Shibahara S, Yoshizawa M, Suzuki H, et al. Functional analysis of cDNAs for two types of human heme oxygenase and evidence for their separate regulation. J Biochem (Tokyo) 1993;113:214–218.
Maines MD . The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997;37:517–554.
McCoubrey Jr WK, Maines MD . The structure, organization and differential expression of the gene encoding rat heme oxygenase-2. Gene 1994;139:155–161.
Tenhunen R, Marver HS, Schmid R . The enzymatic catabolism of hemoglobin: stimulation of microsomal heme oxygenase by hemin. J Lab Clin Med 1970;75:410–421.
Otterbein LE, Choi AM . Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol 2000;279:L1029–L1037.
Ryter SW, Choi AM . Heme oxygenase-1: molecular mechanisms of gene expression in oxygen-related stress. Antioxid Redox Signal 2002;4:625–632.
Immenschuh S, Tan M, Ramadori G . Nitric oxide mediates the lipopolysaccharide dependent upregulation of the heme oxygenase-1 gene expression in cultured rat Kupffer cells. J Hepatol 1999;30:61–69.
Rizzardini M, Carelli M, Cabello Porras MR, et al. Mechanisms of endotoxin-induced haem oxygenase mRNA accumulation in mouse liver: synergism by glutathione depletion and protection by N-acetylcysteine. Biochem J 1994;304:477–483.
Cantoni L, Rossi C, Rizzardini M, et al. Interleukin-1 and tumour necrosis factor induce hepatic haem oxygenase. Feedback regulation by glucocorticoids. Biochem J 1991;279:891–894.
Mitani K, Fujita H, Kappas A, et al. Heme oxygenase is a positive acute-phase reactant in human Hep3B hepatoma cells. Blood 1992;7:1255–1259.
Oguro T, Takahashi Y, Ashino T, et al. Involvement of tumor necrosis factor alpha, rather than interleukin-1alpha/beta or nitric oxides in the heme oxygenase-1 gene expression by lipopolysaccharide in the mouse liver. FEBS Lett 2002;516:63–66.
Rizzardini M, Zappone M, Villa P, et al. Kupffer cell depletion partially prevents hepatic heme oxygenase 1 messenger RNA accumulation in systemic inflammation in mice: role of interleukin 1beta. Hepatology 1998;27:703–710.
Alcaraz MJ, Fernandez P, Guillen MI . Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr Pharm Des 2003;9:2541–2551.
Otterbein LE, Soares MP, Yamashita K, et al. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol 2003;24:449–455.
Wunder C, Potter RF . The heme oxygenase system: its role in liver inflammation. Curr Drug Targets Cardiovasc Haematol Disord 2003;3:199–208.
Ramadori G, Christ B . Cytokines and the hepatic acute-phase response. Semin Liver Dis 1999;19:141–155.
Streetz KL, Wustefeld T, Klein C, et al. Mediators of inflammation and acute phase response in the liver. Cell Mol Biol (Noisy -le-grand) 2001;47:661–673.
Lyoumi S, Puy H, Tamion F, et al. Nitric oxide synthase inhibition and the induction of cytochrome P-450 affect heme oxygenase-1 messenger RNA expression after partial hepatectomy and acute inflammation in rats. Crit Care Med 1998;26:1683–1689.
Fey GH, Fuller GM . Regulation of acute phase gene expression by inflammatory mediators. Mol Biol Med 1987;4:323–338.
Shibahara S, Muller R, Taguchi H, et al. Cloning and expression of cDNA for rat heme oxygenase. Proc Natl Acad Sci USA 1985;82:7865–7869.
Northemann W, Braciak TA, Hattori M, et al. Structure of the rat interleukin 6 gene and its expression in macrophage-derived cells. J Biol Chem 1989;264:16072–16082.
Gehring MR, Shiels BR, Northemann W, et al. Sequence of rat liver alpha 2-macroglobulin and acute phase control of its messenger RNA. J Biol Chem 1987;262:446–454.
Kioussis D, Eiferman F, van de RP, et al. The evolution of alpha-fetoprotein and albumin. II. The structures of the alpha-fetoprotein and albumin genes in the mouse. J Biol Chem 1981;256:1960–1967.
Fort P, Marty L, Piechaczyk M, et al. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res 1985;11 (13):1431–1442.
Barbu V, Dautry F . Northern blot normalization with a 28S rRNA oligonucleotide probe. Nucleic Acids Res 1989;17:7115.
Knook DL, Sleyster EC . Separation of Kupffer and endothelial cells of the rat liver by centrifugal elutriation. Exp Cell Res 1976;99:444–449.
Armbrust T, Nordmann B, Kreissig M, et al. C1Q synthesis by tissue mononuclear phagocytes from normal and from damaged rat liver: up-regulation by dexamethasone, down-regulation by interferon gamma, and lipopolysaccharide. Hepatology 1997;2:98–106.
Laemmli UK . Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–685.
Towbin H, Staehelin T, Gordon J . Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979;76:4350–4354.
Chirgwin JM, Przybyla AE, MacDonald RJ, et al. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 1979;18:5294–5299.
Braissant O, Wahli W . A simplified in situ hybridization protocol using non-radioactively labeled probes to detect abundant and rare mRNAs on tissue sections. Biochemica 1998;1:10–16.
Luheshi GN, Stefferl A, Turnbull AV, et al. Febrile response to tissue inflammation involves both peripheral and brain IL-1 and TNF-alpha in the rat. Am J Physiol 1997;272 (3 Part 2):R862–R868.
Luckey SW, Taylor M, Sampey BP, et al. 4-hydroxynonenal decreases interleukin-6 expression and protein production in primary rat Kupffer cells by inhibiting nuclear factor-kappaB activation. J Pharmacol Exp Ther 2002;302:296–303.
Schreiber G, Aldred AR, Thomas T, et al. Levels of messenger ribonucleic acids for plasma proteins in rat liver during acute experimental inflammation. Inflammation 1986;10:59–66.
Knittel T, Fellmer P, Neubauer K, et al. The complement-activating protease P100 is expressed by hepatocytes and is induced by IL-6 in vitro and during the acute phase reaction in vivo. Lab Invest 1997;77:221–230.
Lyoumi S, Puy H, Tamion F, et al. Heme and acute inflammation role in vivo of heme in the hepatic expression of positive acute-phase reactants in rats. Eur J Biochem 1999;261:190–196.
Fattori E, Cappelletti M, Costa P, et al. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med 1994;180:1243–1250.
Luster MI, Germolec DR, Yoshida T, et al. Endotoxin-induced cytokine gene expression and excretion in the liver. Hepatology 1994;19:480–488.
Ulich TR, Guo KZ, Irwin B, et al. Endotoxin-induced cytokine gene expression in vivo. II. Regulation of tumor necrosis factor and interleukin-1 alpha/beta expression and suppression. Am J Pathol 1990;137:1173–1185.
Ulich TR, Guo KZ, Remick D, et al. Endotoxin-induced cytokine gene expression in vivo. III. IL-6 mRNA and serum protein expression and the in vivo hematologic effects of IL-6. J Immunol 1991;146:2316–2323.
Heinrich PC, Castell JV, Andus T . Interleukin-6 and the acute phase response. Biochem J 1990;265:621–636.
Hoffmann R, Henninger HP, Schulze-Specking A, et al. Regulation of interleukin-6 receptor expression in rat Kupffer cells: modulation by cytokines, dexamethasone and prostaglandin E2. J Hepatol 1994;21:543–550.
Geisterfer M, Richards C, Baumann M, et al. Regulation of IL-6 and the hepatic IL-6 receptor in acute inflammation in vivo. Cytokine 1993;5:1–7.
Kampfer H, Kolb N, Manderscheid M, et al. Macrophage-derived heme-oxygenase-1: expression, regulation, and possible functions in skin repair. Mol Med 2001;7:488–498.
Ohta K, Kikuchi T, Arai S, et al. Protective role of heme oxygenase-1 against endotoxin-induced uveitis in rats. Exp Eye Res 2003;77:665–673.
Song R, Kubo M, Morse D, et al. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol 2003;163:231–242.
Tamion F, Richard V, Bonmarchand G, et al. Induction of heme-oxygenase-1 prevents the systemic responses to hemorrhagic shock. Am J Respir Crit Care Med 2001;164:1933–1938.
Tullius SG, Nieminen-Kelha M, Buelow R, et al. Inhibition of ischemia/reperfusion injury and chronic graft deterioration by a single-donor treatment with cobalt-protoporphyrin for the induction of heme oxygenase-1. Transplantation 2002;74:91–98.
Vicente AM, Guillen MI, Habib A, et al. Beneficial effects of heme oxygenase-1 up-regulation in the development of experimental inflammation induced by zymosan. J Pharmacol Exp Ther 2003;307:1030–1037.
Soares MP, Seldon MP, Gregoire IP, et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol 2004;15 (172):3553–3563.
Wagener FA, Eggert A, Boerman OC, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001;98:1802–1811.
Bussolati B, Ahmed A, Pemberton H, et al. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood 2004;103:761–766.
Poss KD, Tonegawa S . Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA 1997;94:10919–10924.
Bauer I, Wanner GA, Rensing H, et al. Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology 1998;27:829–838.
Goda N, Suzuki K, Naito M, et al. Distribution of heme oxygenase isoforms in rat liver. Topographic basis for carbon monoxide-mediated microvascular relaxation. J Clin Invest 1998;101:604–612.
Kiemer AK, Gerwig T, Gerbes AL, et al. Kupffer-cell specific induction of heme oxygenase 1 (hsp32) by the atrial natriuretic peptide—role of cGMP. J Hepatol 2003;38:490–498.
Song Y, Shi Y, Ao LH, et al. TLR4 mediates LPS-induced HO-1 expression in mouse liver: role of TNF-alpha and IL-1beta. World J Gastroenterol 2003;9:1799–1803.
Bauer I, Rensing H, Florax A, et al. Expression pattern and regulation of heme oxygenase-1/heat shock protein 32 in human liver cells. Shock 2003;20:116–122.
Fernandez M, Bonkovsky HL . Increased heme oxygenase-1 gene expression in liver cells and splanchnic organs from portal hypertensive rats. Hepatology 1999;29:1672–1679.
Mostert V, Hill KE, Ferris CD, et al. Selective induction of liver parenchymal cell heme oxygenase-1 in selenium-deficient rats. Biol Chem 2003;384:681–687.
Morita T, Kourembanas S . Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J Clin Invest 1995;96:2676–2682.
Brune B, Ullrich V . Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol 1987;32:497–504.
Otterbein LE, Bach FH, Alam J, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 2000;6:422–428.
Lee TS, Chau LY . Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med 2002;8:240–246.
Stocker R, Yamamoto Y, McDonagh AF, et al. Bilirubin is an antioxidant of possible physiological importance. Science 1987;235:1043–1046.
Nakagami T, Toyomura K, Kinoshita T, et al. A beneficial role of bile pigments as an endogenous tissue protector: anti-complement effects of biliverdin and conjugated bilirubin. Biochim Biophys Acta 1993;1158:189–193.
Kato Y, Shimazu M, Kondo M, et al. Bilirubin rinse: A simple protectant against the rat liver graft injury mimicking heme oxygenase-1 preconditioning. Hepatology 2003;38:364–373.
Ferris CD, Jaffrey SR, Sawa A, et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol 1999;1:152–157.
Baranano DE, Wolosker H, Bae BI, et al. A mammalian iron ATPase induced by iron. J Biol Chem 2000;275:15166–15173.
Eisenstein RS, Garcia-Mayol D, Pettingell W, et al. Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc Natl Acad Sci USA 1991;88:688–692.
Acknowledgements
This work was supported by Grants SFB 402 TP C6, SFB 402 TP D3 and GRK 335 from the Deutsche Forschungsgemeinschaft. This paper is dedicated to Professor Karl Hermann Meyer zum Bueschenfelde for his 75th birthday.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tron, K., Novosyadlyy, R., Dudas, J. et al. Upregulation of heme oxygenase-1 gene by turpentine oil-induced localized inflammation: involvement of interleukin-6. Lab Invest 85, 376–387 (2005). https://doi.org/10.1038/labinvest.3700228
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700228
Keywords
This article is cited by
-
Induction of chemokines and cytokines before neutrophils and macrophage recruitment in different regions of rat liver after TAA administration
Laboratory Investigation (2014)
-
Ferroportin-1 is a ‘nuclear'-negative acute-phase protein in rat liver: a comparison with other iron-transport proteins
Laboratory Investigation (2012)
-
Comparison of changes in gene expression of transferrin receptor-1 and other iron-regulatory proteins in rat liver and brain during acute-phase response
Cell and Tissue Research (2011)
-
Changes in gene expression of DOR and other thyroid hormone receptors in rat liver during acute-phase response
Cell and Tissue Research (2010)
-
Low serum selenium is associated with anemia among older adults in the United States
European Journal of Clinical Nutrition (2009)
