
Abstract
Preinvasive mammary pathologies in humans and rat chemical carcinogenesis model systems have an increased microvascular density relative to normal tissue. This suggests the possibility of preventing invasive breast cancer by inhibiting angiogenesis. Vascular endothelial cell growth factor (VEGF) is a potent angiogenic growth factor, commonly involved in tumor-induced angiogenesis. Here, we show that both VEGF and VEGFR2 expression increase with histological progression to invasive disease in the rat 7,12-dimethylbenz[a]anthracene (DMBA) model. Other VEGF receptors, VEGFR1, neuropilin 1 and neuropilin 2, are constitutively expressed throughout progression. To examine whether VEGF signaling is functionally relevant to tumor-induced endothelial tubule formation in vitro and for tumor formation in vivo, we utilized the VEGFR2 inhibitor, ZD6474. In vitro endothelial cell tubulogenesis induced by isolated mammary organoids or carcinoma in situ from DMBA-treated rats is inhibited by ZD6474, in a dose-dependent fashion. The administration of ZD6474 to DMBA-treated rats inhibits the formation of atypical ductal hyperplasia and carcinoma in situ by greater than 95% (P<0.05), when administered 1 week or 6 weeks post-DMBA initiation. Invasive disease was absent in all ZD6474 cohorts. These data support the hypothesis that progression of DMBA-induced preinvasive mammary pathologies to palpable disease requires angiogenesis via a VEGF-dependent mechanism.
Similar content being viewed by others


Main
Angiogenesis is the process of forming new vessels from pre-existing vessels,1 and has been shown to be important in the process of breast cancer progression and metastasis.2 We and others have shown that angiogenesis is also important in the process of breast cancer formation, as indicated by the step-wise increase in microvascular density with progression from normal epithelium to invasive disease.3, 4, 5, 6 In addition, we have shown that in the rat model system of DMBA-induced mammary tumor formation there is a similar pattern of upregulated microvascular density with progression.7 Finally, if one isolates normal epithelium or tumor tissue from DMBA-treated rats, both are capable of inducing endothelial tubule formation in vitro, indicating that the angiogenic potential of these tissues is greater than that from vehicle-treated control rats.7
One of the clinical problems in caring for women with breast disease is determining how to prevent cancer formation in women who have increased risk due to genetic predisposition or the presence of proliferative lesions.8 The anti-estrogen, tamoxifen, is currently the best-studied option for chemoprevention in this population.9 Data regarding increasing microvascular density with disease progression suggests that inhibition of angiogenesis may be a novel mechanistic target for chemoprevention. As proof-of-principle that disease progression is dependent upon angiogenesis, we examined the effect of the angiogenic inhibitor, TNP-470, on disease progression in carcinogen-treated rats. These data showed that early administration of this endothelial-selective inhibitor potently blocked the production of carcinoma in situ (CIS) and invasive disease (INV).10 Our early characterization of DMBA-induced tumors showed that they produced the potent angiogenic growth factor, vascular endothelial cell growth factor (VEGF).7 Here, we examine the expression levels of VEGF and its receptors, VEGFR1, VEGFR2, Neuropilin-1 (NRP1), and Neuropilin-2 (NRP2), during DMBA-induced carcinogenesis in rats. In addition, we show potent effects of the VEGFR2 inhibitor, ZD6474, at very early stages of the carcinogenic process in this model.
Materials and methods
Animal Model
Virgin female Sprague–Dawley rats (Taconic Farms, Germantown, NY, USA), 45 days old, were given 8 mg DMBA/100 g body weight (16 mg/ml in corn oil dissolved at 95–96°C). DMBA-treated rats were randomized to three cohorts (n=17 per cohort). Cohort A were controls, receiving 1% Tween-80 (Sigma, St Louis, MO, USA) by intragastric gavage daily under light isofluorane anesthesia. Cohort B were given a daily gavage of ZD6474 (gift from Astra Zeneca) for a period of 12 weeks beginning on day 2 (30 mg/kg weeks 1–6, 15 mg/kg weeks 7–12). Cohort C were given daily gavage of ZD6474 for a period 6 weeks (30 mg/kg, weeks 7–12). ZD6474 was administered as a suspension in 1% Tween-80 as described for controls. Animals were palpated twice weekly for the presence of tumors. BrdU (50 mg/kg) was administered i.p. to all animals at 24 and 12 h prior to death. At 12 weeks after DMBA treatment, mammary glands, including tumors and hyperplastic patches, were dissected for assays, snap frozen for RNA extraction, or formalin fixed for histological analysis. Pathologic diagnoses were according to Russo et al11 and read blinded to treatment group. We have recently published a pictoral description of diagnoses, as used in these studies.10 All cases of INV were confirmed with cytokeratin immunohistochemistry. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Immunohistochemistry
Immunohistochemistry was performed using the Ventana 320ES automatic immunostaining system, with primary and secondary antibody incubations for 32 min at room temperature, as described previously.7 For the monoclonal primaries rat adsorbed anti-mouse (Vector, Burlingame, CA, USA) was used as the secondary antibody at 1:50. Preimmune rabbit IgG or isotype-specific mouse IgG added in place of the primary antibody were used as the negative controls. Histologic sections were double stained by immunohistochemistry for von Willebrand factor (VWF) (polyclonal; 1:400, Dako, Carpenteria, CA, USA) and bromodeoxyuridine (BrdU) (clone 3D9, 1:500, which was a gift from Dr Azra Raza, Rush Presbyterian Hospital, Chicago, IL, USA). In addition, cytokeratin (clone C-11, 1:20, Lab Vision, Fremont, CA, USA) staining was performed on tumors that were potentially invasive. Slides for cytokeratin staining were pretreated in 10 mM borate at 75°C overnight prior to the addition of the primary antibody.
TUNEL Labeling
Deparaffinized slides were labeled to identify apoptotic cells using the TUNEL labeling kit from Roche (Indianapolis, IN, USA) according to the manufacturer's instructions.
RT-PCR
Frozen sections of rat mammary glands 3 months post-DMBA exposure were used for RT-PCR. Alternatively, the PixCell II LCM system was used to microdissect epithelial cells from H&E-stained frozen tissue sections. Amplitude and pulse duration of the PixCell II laser were adjusted to allow complete tissue capture using 15 μm laser beam. The microdissecting time of each section was within 15 min to prevent RNA degradation. After microdissection of each specimen, the thermoplastic film-coated cap containing the captured cells was placed in a microtube. Isolated RNA (Stratagene RNA Micro-isolation Kit, Stratagene, Carlsbad, CA, USA) was resuspended in DEPC water and reverse transcribed to cDNA using a cDNA Cycle Kit (Stratagene) according to the manufacturer's instructions. PCR was performed on cDNA using primers listed below and the following: 1 μl of cDNA, 2 μl 10 × buffer, 2 μl 5 pmol primers, 0.4 μl 10 nM dNTP, and 0.3 μl Taq in 20 μl. PCR conditions included hot start at 94°C for 3 min, 30 s denaturation (94°C), 30 s annealing (annealing temperatures (AT) listed below), and 30 s extension (72°C) for 25–35 amplification rounds and 5 min final extension at 72°C. PCR reaction mix (5 μl) was separated by PAGE using 5% acrylamide gel with SYBR Green I staining. Gels were scanned with STORM 860 (Molecular Dynamics, Inc.) and the intensity of PCR product bands was quantified using ImageQuanT software. The results are expressed as the ratio of VEGF or receptor band intensity/β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity. Note that one of two actin primer pairs were used, depending on the product size (PS) needed.
Data from semi-quantitative RT-PCR was confirmed for VEGFR2 using real-time PCR, comparing product formation with GAPDH as a standard. Real-time PCR was performed using a LightCycler® (Smart Cycler System.II, Cepheid, CA, USA). Reactions were done in a final volume of 30 μl using primers 0.5 μM (as above), 1 μl synthesized cDNA, dNTPs, and Taq DNA polymerase and reaction buffer provided in the LightCycler-FastStart DNA Master SYBR Green I kit (Roche Molecular Biochemicals, Mannheim, Germany). All real-time PCR assays included an initial 10 min incubation at 95°C to activate the FastStart polymerase enzyme followed by 55 cycles of denaturation for 10 s at 95°C, annealing 6 s at 58°C (for GAPDH) or as indicated for each primer and extension 10 s at 72°C. Melting curves were plotted to determine PCR product identity. Results were analyzed using Smart Cycler software.
Primers are as follows:
-
VEGF (GI: 6708499) AT: 65°C; PS: 145 bp
-
Sense: 5′-GCTCTCTTGGGTGCACTGGAC-3′
-
Antisense: 5′-ACGGCAATAGCTGCGCTGGTA-3′
-
VEGFR-112 (GI: 511662) AT: 58°C; PS: 240 bp
-
Sense: 5′-CAAGGGACTCTACACTTGTC-3′
-
Antisense: 5′-CCGAATAGCGAGCAGATTTC-3′
-
VEGFR-212 (GI: 2098758) AT: 60°C; PS: 537 bp
-
Sense: 5′-GCCAATGAAGGGGAACTGAAGA-3′
-
Antisense: 5′-CTCTGACTGCTGGTGATGCTGTC-3′
-
NPR1 (GI: 2367638) AT: 58°C; PS: 333 bp
-
Sense: 5′-CCACAGAGAAGCCAACCATT-3′
-
Antisense: 5′-TGACCCTCAGTGTACCCACA-3′
-
NPR2 (GI: 2367640) AT: 58°C; PS: 320 bp
-
Sense: 5′-ACGTGGATTCCAACAAGGAG-3′
-
Antisense: 5′-GCAACCAAAGAGCTCCAGTC-3′
-
EGFR13 (GI: 6478867) AT: 56°C; PS: 457 bp
-
Sense: 5′-GTACTGCACTGCCATCAGTG-3′
-
Antisense: 5′-CCGAGGAGCATAAAGGATTA-3′
-
β-actin (GI: 5016088) AT: as per other primer; PS: 223 bp
-
Sense: 5′-CGCGAGAAGATGACCCAGATCATG-3′
-
Antisense: 5′-AGGATCTTCATGAGGTAGTCAGT-3′
-
or β-actin purchased from Stratagene; AT: as per other primer; PS: 661 bp
-
Sense: 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′
-
Antisense: 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′
-
GAPDH14 (GI: 204248) AT: 56°C; PS: 294 bp
-
Sense: 5′-GGTCATCCCTGAGCTGAACG-3′
-
Antisense: 5′-TCCGTTGTCATACCAGGAAAT-3′
In Vitro Endothelial Tubulogenesis (Angiogenic Potential)
As previously described,7 fresh minced tumor was placed over the surface layer of a collagen type I sandwich, 1.5 mg/ml (rat-tail collagen), in which 2 × 104 HUVEC (Clonetics, San Diego, CA, USA) per well had been cultured in between collagen layers of a 48-well plate. Tissue pieces for each well were isolated under 10 × visualization to assure an equal distribution. Five wells per condition were used for each test reagent. Alternatively, viably frozen minced tumor pieces are thawed and cultured in type 1 collagen in DMEM/F12 (50:50) with 12 mM Hepes, 10 μg/ml insulin, 1 mg/ml BSA, 1 μg/ml ascorbic acid, 0.5 mM glutamine, 0.5 μg/ml hydrocortisone, 2.6 ng/ml selenium, 1 μg/ml prolactin, 1 μg/ml progesterone, 10 ng/ml apotransferin, and 10 ng/ml EGF. 24 h conditioned media is concentrated in a Centricon YM-3 (Millipore Corp, Bedford, MA, USA) and used to stimulate HUVEC tubulogenesis. Monoclonal anti-human VEGF (clone JH121, Neomarkers, Fremont, CA, USA) or mouse immunoglobulin (Sigma) were used at 1, 5, 10 and 20 μ g/ml. ZD6474 (a gift from AstraZeneca) was added at final concentrations between 0 and 500 nM. bFGF or VEGF (75 ng/ml) (NCI) were used as positive controls. In some assays, EGF or TGFα (10 ng/ml) (BD Biosciences, Bedford, MA, USA) were used to induce angiogenesis. After 15 h, 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (1 mg/ml) (Sigma) was added. After 2 h later the medium was removed and the wells fixed in 10% formalin. If tissue pieces were used, these were removed and checked for viability and equality of distribution among replicate wells by MTT staining. Digital images of five replicate wells were analyzed for total length of HUVEC tubules using ImagePro (Media Cybernetics) and normalized against medium alone and medium with 75 ng/ml VEGF165.
In Vitro Endothelial Mitogenesis
Conditioned medium from DMBA-induced tumors in collagen type I cultures (as above) were used to induce HUVEC proliferation. The medium was first concentrated using a Centricon YM-3 (Millipore Corp, Bedford, MA, USA). The concentrate was added to 8 × 103 HUVEC per well in 48-well plates. ZD6474 was added at 0, 100, 250, and 500 nM. Five replicates were performed per condition per sample. After 40 h the HUVEC were stained with 5 mg/ml MTT. After 2 h, cells were dissolved in DMSO and the optical density recorded at 550 nm.
Statistical Methods
All statistical evaluations were performed using Sigma Stat (SPSS, Chicago, IL, USA). Group mean comparisons were by one-way ANOVA, two-sided, with 95% confidence intervals. Multiple comparisons within one-way ANOVA were by Bonferroni t-test. PCR product ratios were compared using two-sided nonparametric ANOVA on ranks with 95% confidence intervals. Matched comparisons at a given concentration of immunoglobulin were evaluated by T-test.
Results
Mammary glands and tumors were isolated and snap frozen in OCT from rats 3 months following DMBA exposure. Extracted RNA from frozen tissue sections were used for semi-quantitative analysis of VEGF, VEGFR1, VEGFR2, NRP1, and NRP2 expression relative to the expression of β-actin (Figure 1). Seven to 15 samples for each histological diagnosis were examined. Note that VEGF expression increases with disease progression, as does the expression of VEGFR2. Expression levels of VEGFR1 and the VEGF coreceptors, NRP1 and NRP2, do not differ among the different pathologies examined. To confirm the VEGFR2 data, the studies were repeated using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for normalization. The same relative expression patterns were observed as with β-actin (Figure 1). Finally, cDNA samples from five normal, four IDPp, five CIS, and four INV were analyzed by real-time PCR relative to GAPDH, yielding similar results (data not shown). ZD6474 has a 12.5-fold increased selectivity for VEGFR2 over the epidermal growth factor receptor (EGFR) with an IC50 for EGFR of 0.5±0.1 μM;15 therefore, we also examined EGFR mRNA expression during tumorigenesis (data not shown). EGFR was expressed at very low levels in normal tissue and did not increase with progression, consistent with previously published data showing that the EGFR ligand, transforming growth factor α, but not EGFR, are found in DMBA-induced tumors.16

(a) Mean±s.d. of RT-PCR product levels quantified by phosphoimager analysis are shown. Data are expressed as the band intensity for each growth factor or receptor listed relative to the actin or GAPDH level determined in the same PCR reaction. Means are derived from the combined results of 7–15 specimens per histological type. (a) *Indicates statistical significance relative to normal tissue from DMBA-treated rats (P<0.05). (b) Examples of gels used in the analysis for panel (a) are shown, where M=size marker, Normal=normal mammary tissue from DMBA-treated rats.
Since tumor cell VEGF/VEGFR2 coexpression may form an autocrine loop in some systems,17 we tried to confirm that expression of VEGFR2 was restricted to the endothelium. Antibodies specific to VEGF/VEGFR2 complexes did not bind specifically in our rat tissues. RT-PCR for VEGFR2 from isolated epithelial organoids showed VEGFR2 expression in three of 11 samples (data not shown), indicating a rare vascular contamination in these few isolates and no expression in the majority of samples. To examine the epithelium of preinvasive lesions, we isolated epithelial cells from frozen tissue sections using laser capture microdissection. Samples were matched to those used for RT-PCR of whole tissue sections, above. Product production was normalized to GAPDH in the same tube. Figure 2 shows that compared to whole tissue sections containing IDPp or CIS, there is little to no production of VEGFR2 in the epithelium from these matched lesions.

Three CIS and three IDPp samples from the analyses in Figure 1 were chosen for examination of the epithelium by lasar capture microdissection. Individual microdissected samples were assayed by RT-PCR and the ratio to GAPDH determined, as above. Shown are measurements from individual RT-PCR reactions. Note for IDPp that while the tissue sections always gave a correct reaction product, two of the samples of IDPp microdissected epithelium showed no expression of VEGFR2, despite a robust expression of GAPDH.
To confirm that the VEGF produced by the tumors was sufficient to drive angiogenesis, we assayed the ability of anti-VEGF antibody or the VEGFR2 inhibitor, ZD6474, to inhibit tissue-induced endothelial tubule formation in vitro. As previously shown, isolated mammary organoids from DMBA-treated rats and fragments of CIS diced to pieces 0.5–1 mm on each side readily induce tubule formation in HUVEC embedded in collagen gels.7 Concentrated conditioned media from CIS in culture is also able to induce HUVEC tubulogenesis. Figure 3 shows the mean ±s.d. of HUVEC tubule length induced by three tumors in which 0–20 μg/ml IgG or anti-VEGF was added to the angiogenesis assay. In all three tumors 10 and 20 μg/ml of anti-VEGF showed statistically significant inhibition relative to the same concentration of nonspecific mouse immunoglobulin (P<0.05).

Plotted is the mean±s.d. of data from three separate tumors. HUVEC tubulogenesis was stimulated using conditioned medium from three tumors assayed in five replicate wells for each Ig condition. Relative tubule length is the total HUVEC tubule length per microscopic field area. Either nonspecific immunoglobulin or anti-VEGF were added to the assay at the concentrations indicated. *Indicates statistically significant differences by t-test between the two treatment conditions at that concentration (P<0.05).
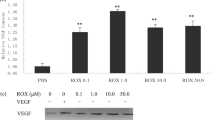
ZD6474 was able to inhibit tumor-induced angiogenesis in seven of nine tumors tested, in a dose-dependent manner. Combined data from all nine tumors shows that ZD6474 inhibits tumor-induced HUVEC tubule formation in a dose-dependent fashion above concentrations of 250 nM (Figure 4a). Similar data are also seen when normal organoids from DMBA-treated rats are utilized, although a slightly higher concentration (500 nM) was necessary in these assays to reach statistical significance (Figure 4b). HUVEC tubulogenesis is an in vitro test of angiogenic potential. A second test is endothelial proliferation. Figure 4c shows that ZD6474 also blocks tumor conditioned medium-induced HUVEC proliferation in a dose-dependent fashion. As with most tumors, DMBA-induced tumors produce a variety of angiogenic factors;18 therefore, to ensure the specificity of ZD6474 for VEGF-mediated signaling, we tested this agent against basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), transforming growth factor alpha (TGFα), and VEGF-induced HUVEC tubulogenesis (Figure 4c). EGF and TGFα were chosen since they are angiogenic factors, but also ligands to EGFR, another receptor tyrosine kinase against which ZD6474 has some activity.15 Note that when growth factors other than VEGF are used as the angiogenic agent, ZD6474 has no effect on HUVEC tubulogenesis, whereas ZD6474 gives significant inhibition of VEGF-stimulated tubules under the same conditions (P<0.05).

HUVEC tubulogenesis assays were performed as in Figure 3. Wells contained either isolated DMBA-induced carcinoma (a) or mammary organoids from DMBA-treated rats (b). ZD6474 was added during the assay at the concentrations indicated. Tumor data from nine specimens (a) or organoid data from five specimens (b) were pooled for statistical analysis by normalizing the HUVEC tubule length for each set of wells to a minimum tubule length (zero) and VEGF-induced tubule length (100%). As an additional assay to show the VEGFR2-dependent effects of tumors on the endothelium, we used conditioned media from three of the tumors to induce endothelial cell proliferation. MTT-stained HUVEC were dissolved in DMSO and the OD determined at 550 nm. The inhibitory effect of ZD6474 is shown in (c). The mean±s.d. for each type of assay are indicated. *Indicates a statistically significant difference relative to 0 nM ZD6474 (P<0.05). To confirm the specificity of ZD6474 for VEGFR2 in our assay system, HUVEC were treated with 10 ng/ml EGF, 10 ng/ml TGFα, 75 ng/ml bFGF, or 75 ng/ml VEGF and ZD6474, as shown (d). Plotted is the mean±s.d. from two sets of experiments, each with 5 replicate wells per treatment. *Indicates statistically different results relative to growth factor alone by ANOVA (P<0.05).
To test the hypothesis that VEGF is a major regulator of vessel growth during DMBA-induced tumor formation and to determine at which stage VEGF regulation of angiogenesis may be important for progression, we administered the VEGFR2 inhibitor, ZD6474, to DMBA-treated rats. ZD6474 is an orally administered low molecular weight inhibitor of VEGFR2 tyrosine kinase activity.15 As in previous studies,10 two time points post-DMBA exposure were chosen to begin drug administration: first, 24 h after DMBA exposure, the time point at which DMBA-DNA adduct formation is largely completed, but before any histological changes in the mammary tissue are identified (Cohort B); second, 6 weeks post-DMBA exposure, a time point when numerous small proliferative lesions, as well as small carcinomas in situ, are readily found (Cohort C). Cohort A received vehicle rather than ZD6474 beginning 24 h after DMBA. All animals were killed 3 months after DMBA exposure and the mammary glands characterized histologically for number of proliferative ductal lesions, CIS, and INV. Figure 5a shows the results of these studies. The incidence data for proliferative lesions are plotted for patches of initiated intraductal proliferations (IDPi) and promoted intraductal proliferations (IDPp) showing cytological atypia, both of which are precursors to CIS and INV. Histological characterization of these lesions and their progression over time have been well described and illustrated, previously.10, 19 In addition, data are shown on lobular hyperplasia (HP), which are precursors to nonaggressive tubular adenomas.20 In this model system all forms of hyperplasia have a greater microvascular density than normal epithelium, but IDPp have the greatest microvascular area relative to epithelial area of any of the DMBA-induced pathologies.7 Indeed, IDPp formation was potently inhibited by ZD6474 when administered early in tumorigenesis, being inhibited by 99% (P<0.05). CIS was similarly inhibited (100%, P<0.05). When administered after the formation of proliferative lesions and small CIS, ZD6474 inhibited IDPp and CIS by 95% (P<0.05). The data on INV did not reach statistical significance in either cohort because in this particular trial, fewer of the tumors progressed to invasion in the DMBA group relative to previous studies. However, in the ZD6474 cohorts there were no invasive lesions identified.

(a) DMBA-treated rats were randomized to three cohorts, as described in Materials and Methods. Cohort A was vehicle controlled. Cohort B was given a daily gavage of ZD6474 for a period of 12 weeks beginning on day 2. Cohort C was given a daily gavage of ZD6474 for a period of 6 weeks beginning at week 7 after DMBA treatment. Plotted is the mean±s.d. number of each pathology per rat for each group, determined 12 weeks post-DMBA exposure. *Indicates a P value of <0.05 relative to the Cohort A by one-way analysis of variance. (b) During the course of the study, rats were palpated twice weekly for tumor burden. Plotted is the number of tumors in each cohort by week. No tumors were found in Cohort B, so these data are omitted. (c) Plotted are the volumes of tumors palpated per cohort over time.
Histological examination of the rare tumors in the ZD6474 cohort showed tumors that were in part necrotic, with large viable regions lacking significant epithelial proliferation (Figure 6b) relative to tumors from the untreated DMBA cohort (Figure 6a). Tumors shown are double labeled for von Willebrand Factor (brown) and BrdU (blue). Note that the nonproliferative area in panel b is not necrotic but contains only rare BrdU-positive cells. Vessels are indicated by arrows. Vessels in the untreated group appear normal; whereas the vessels in the ZD6474 cohort are often ill-defined and surrounded by spilling of plasma proteins into the adjacent matrix. Three tumors each from the untreated and the 6-week ZD6474 cohorts were examined by image analysis for vascular area relative to epithelial area. DMBA-induced CIS had a normalized vascular area of 0.087±0.122 vs 0.023±0.005 in the ZD6474-treated cohort. There are too few tumors for statistical analyses, but the data suggest that unlike TNP-470, which did not alter the normalized vascular area,10 ZD6474 breakthrough tumors do have a diminished vascularity.

Panels (a) and (b) show histological sections of CIS from the untreated and 6-week ZD6474 cohorts, respectively. The tissue are stained by immunohistochemistry for von Willebrand Factor (brown) and BrdU (blue). Vessels are indicated by arrows. Both are × 40 magnification with the micron bar shown in panel (a) Panels (c) and (d) show histological sections from the same tumors stained by TUNEL. Apoptotic cells and clusters of cells are indicated by arrows. Both are × 100 magnification with the micron bar shown in panel (c).
Figure 6c and d show the same tumors as in panels a and b now stained for apoptosis by TUNEL assay. Brown apoptotic cells are highlighted by arrows. The apparent apoptotic rate from these images suggests that the ZD6474 cohort had tumors with a decreased apoptotic frequency relative to the untreated cohort; however, there are too few available tumors to make a firm conclusion.
The efficacy of ZD6474 as a chemopreventive agent is most apparent when one considers palpation data (Figure 5b). In the early cohort (Cohort B; data not shown) there were no palpable tumors in the ZD6474-treated cohort compared with a mean tumor volume of 14 cm3/rat in the vehicle-treated control group (Cohort A, Figure 5c). In addition, the effect of ZD6474 was readily apparent in the 6-week group (Cohort C) in which many of the animals had small palpable tumors prior to the initiation of therapy. However, most disappeared within 3 weeks of therapy. In some cases, IDP were identified in these locations. In other cases, histological confirmation of a previous tumor at that site was not possible.
Discussion
It has been understood for some time that invasive tumor growth and metastasis are angiogenesis dependent. In a few types of tumors, this knowledge has led to some remarkable data in clinical trials and great hope for certain patients.21 However, the role of angiogenesis in preinvasive lesions is much less well understood. In the DMBA model system, by RT-PCR or immunohistochemistry (data not shown), VEGF expression is ubiquitous but rising with disease progression. However, the greatest microvascular density relative to epithelial area is found in IDPp.7 VEGF expression levels do not clearly predict these changes in microvascular density. Therefore, we examined the expression of VEGF receptors. Most studies have shown that VEGFR2 is the receptor commonly associated with angiogenesis, particularly in tumors.22 However, the coreceptors NRP1 and NRP2 increase VEGFR2 signaling in the presence of VEGF.23 VEGFR1 also forms complexes with neuropilins, but the significance of these associations and the role of VEGFR1 in tumor angiogenesis are not clear.24 However, NRP1 has been shown to regulate angiogenesis in wound healing.25 Our data show that all four receptors are ubiquitous throughout progression, but only VEGFR2 expression changes with tumor development. Interestingly, the increase in VEGFR2 levels with the formation of IDPp, seen in many lesions, is consistent with the observed large increase in microvascular density at this stage. The functional inhibition of VEGFR2 by ZD6474 resulted in the suppressed development of microscopic and palpable tumors, showing the centrality of this receptor in tumor-induced angiogenesis. Whether VEGFR1 and the neuropilins also play a role requires additional study.
The inhibition of atypical hyperplasia, IDPp, is surprising if one considers that these microscopic lesions are not predicted to be dependent upon angiogenesis based upon data showing that tumors in perfused organs are limited to 1–2 mm3 prior to neovascularization.1 The maximum diameter of an IDPp in our studies is about 700 μm, which is 0.35 mm3. Therefore, the mechanism by which angiogenesis fosters their growth is not clear. As shown before, IDPp are intensely angiogenic relative to epithelial area, whereas the early hyperplastic lesions (IDPi) have essentially the same level of vascularization as normal epithelium.7 The latter are unaffected by inhibition of VEGF signaling by ZD6474. Although we have not entirely ruled out the possibility of autocrine signaling via this receptor in the epithelium, the levels of VEGFR2 expression make this seem unlikely.
Another well-characterized system in which the relationship of angiogenesis to tumorigenesis has been studied is the Rip-1-Tag islet carcinogenesis system,26 in which a subset of hyperplastic islets stochastically ‘switch on’ angiogenesis followed by progression to CIS and INV. Using this system, Bergers et al27 showed that there was a precursor stage-dependent efficacy of four different angiogenic inhibitors, none of which were able to prevent histological progression to invasive disease. The DMBA-induced pathologies have a different pattern of angiogenic regulation than the Rip-1-Tag model, in that even the normal epithelium is angiogenic relative to that in control rats, with microvascular density increasing during the development of atypical hyperplasia.7 With rapid epithelial expansion to CIS, the microvascular density normalized to epithelial area actually decreases relative to the proliferative lesions. As with human breast disease and unlike the Rip-1-Tag model, vascular disruption and hemorrhage are not a feature of this system. Data presented here show that VEGF expression is ubiquitous in this system, with the highest expression correlating with palpable tumor formation. Therefore, the regulatory step for VEGF-mediated angiogenesis includes upregulation of the receptor, specifically VEGFR2. Expression of this receptor is increased in the late proliferative stage (IDPp), which is consistent with the finding that inhibition of this receptor in vivo by ZD6474 blocks the development of IDPp.
The question then remains, which step in progression is angiogenesis dependent? As in the Rip-1-Tag model,27 the answer depends upon the angiogenic inhibitor examined. In other studies treatment with the angiogenic inhibitor, TNP-470, inhibited CIS and INV formation in this same rat model when administered early, but not at 6 weeks post-DMBA.10 TNP-470 had no influence on ductal hyperplasias, which are precursors to INV, but effectively inhibited lobular hyperplasia. These data are in marked contrast to our current data in which ZD6474 was highly efficacious as a chemopreventive agent when administered early and 6 weeks post-DMBA. Unique to these ZD6474 studies, the atypical ductal proliferations (IDPp) were largely prevented from developing, whereas lobular hyperplasia was unaffected. ZD6474 was even more efficacious than TNP-470 in inhibiting palpable tumor formation and, in addition, was able to block the formation of atypical hyperplasia.
These data add to the growing literature that preinvasive lesions are dependent upon neovascularization for their development.28 Immunohistochemical analysis of angiogenic regulators in human breast pathologies highlights the complexity of angiogenic regulation with progression.29 While there are large numbers of data published in model systems touting novel regulators of an ‘angiogenic switch’, for example,30, 31, 32, 33 it seems far more likely that in human disease there are complex opposing systems that gradually lead to an increased angiogenic phenotype as a function of epithelial progression and stromal response. We have shown this for breast cancer,3 others have shown this for colon cancer.34 This opens up new avenues of research for targeted prevention of invasive tumors. Among populations at risk, those with previously treated malignancies, high-risk family histories, and the presence of preinvasive pathologies are a growing cohort in oncology practices. Our data suggest that chronic inhibition of angiogenesis early in disease may provide new therapeutic modalities in this population of women.
References
Folkman J . What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990;82:4–6.
Weidner N, Semple J, Welch W, et al. Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med 1991;324:1–8.
Heffelfinger S, Yassin R, Miller M, et al. Vascularity of proliferative breast disease and carcinoma in situ correlates with histologic features. Clin Cancer Res 1996;2:1873–1878.
Guidi A, Fischer L, Harris J, et al. Microvessel density and distribution in ductal carcinoma in situ of the breast. J Natl Cancer Inst 1994;86:614–619.
Ottinetti A, Sapino A . Morphometric evaluation of microvessels surrounding hyperplastic and neoplastic mammary lesions. Breast Cancer Res Treat 1988;11:241–248.
Engels K, Fox S, Whitehouse R, et al. Distinct angiogenic patterns are associated with high-grade in situ ductal carcinomas of the breast. J Pathol 1997;181:207–212.
Heffelfinger SC, Gear RB, Taylor K, et al. DMBA-induced pre-invasive mammary tumors are angiogenic in vitro and in vivo. Lab Invest 2000;80:485–492.
Shaaban A, Sloane J, West C, et al. Histopathologic types of benign breast lesions and the risk of breast cancer: case–control study. Am J Surg Pathol 2002;26:421–430.
Clemons M, Danson S, Howell A . Tamoxifen (“Nolvadex”): a review. Cancer Treat Rev 2002;28:165–180.
Heffelfinger S, Gear R, Schneider J, et al. TNP-470 inhibits DMBA-induced mammary tumor formation when administered prior to the formation of cis but is not additive with tamoxifen. Lab Invest 2003;83:1001–1011.
Russo J, Gusterson B, Rogers A, et al. Biology of disease: comparative study of human and rat mammary tumorigenesis. Lab Invest 1990;62:244–278.
Kanellis J, Paizis K, Cox A, et al. Renal ischemia–reperfusion increases endothelial VEGFR-2 without increasing VEGF or VEGFR-1 expression. Kidney Int 2002;61:1696–1706.
Wu S, Sun H, Tu Z, et al. Inhibition of cultured rat prostatic epithelial cell growth by epristeride in vitro. Acta Phamacol Sin 2001;22:257–263.
Goodwin L, Karabinus D, Pergolizzi R, et al. L-type voltage-dependent calcium channel alpha-1C subunit mRNA is present in ejaculated human spermatozoa. Mol Hum Reprod 2000;6:127–136.
Wedge S, Ogilvie D, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res 2002;62:4645–4655.
Ruggeri B, Klurfeld D, Kritchevsky D, et al. Steady-state mRNA expression for growth factors in DMBA-induced rat mammary tumors. Cancer Lett 1990;55:89–93.
Kumar S, Witzig T, Timm M, et al. Expression of VEGF and its receptors by myeloma cells. Leukemia 2003;17:2025–2031.
Yan M, Schneider J, Gear R, et al. Expression of angiogenic factors is upregulated in DMBA-induced rat mammary pathologies. Pathobiology 2004, (in press).
Russo J, Russo I, van Zwieten M, et al. Classification of neoplastic and nonneoplastic lesions of the rat mammary gland. In: Integument and Mammary Glands, Jones TC, Mohr U, Hunt RD (eds). Springer-Verlag: Berlin, 1989, pp 275–304.
Russo J, Russo I . Biology of disease: biological and molecular bases of mammary carcinogenesis. Lab Invest 1987;57:112–137.
Rajkumar S, Gertz M, Kyle R, et al. Mayo Clinic Myeloma A, and Dysproteinemia Group. Current therapy for multiple myeloma. Mayo Clin Proc 2002;77:813–822.
Shinkaruk S, Bayle M, Lain G, et al. Vascular endothelial cell growth factor (VEGF), an emerging target for cancer chemotherapy. Curr Med Chem Anti-Cancer Agents 2003;3:95–117.
Soker S, Takashima S, Miao H, et al. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform specific receptor for vascular endothelial growth factor. Cell 1998;92:735–745.
Fuh G, Garcia K, de Vos A . The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem 2000;275:26690–26695.
Matthies A, Low Q, Lingen M, et al. Neuropilin-1 participates in wound angiogenesis. Am J Pathol 2002;160:289–296.
Hanahan D . Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985;315:115–122.
Bergers G, Javaherian K, Lo K-M, et al. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 1999;284:808–812.
Sharma R, Harris A, Dalgleish A, et al. Angiogenesis as a biomarker and target in cancer chemoprevention. Lancet Oncol 2001;2:726–732.
Heffelfinger S, Miller M, Yassin R, et al. Angiogenic growth factors in pre-invasive breast disease. Clin Cancer Res 1999;5:2867–2876.
Wu M, Tzeng C, Wu L, et al. Thrombospondin-1 acts as a fence to inhibit angiogenesis that occurs during cervical carcinogenesis. Cancer J 2004;10:27–32.
Kitadai Y, Onogawa S, Kuwai T, et al. Angiogenic switch occurs during the precancerous stage of human esophageal squamous cell carcinoma. Oncol Rep 2004;11:315–319.
Chang S, Liu C, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA 2004;101:591–596.
Mobius C, Stein H, Becker I, et al. Athe ‘angiogenic switch’ in the progression of Barrett's metaplasia to esophageal adenocarcinoma. Eur J Surg Oncol 2003;29:890–894.
Shpitz B, Gochberg S, Neufeld D, et al. Angiogenic switch in earliest stages of human colonic tumorigenesis. Anticancer Res 2003;23:5153–5157.
Acknowledgements
The authors would like to acknowledge the gift of ZD6474 from Astra Zeneca. This work was supported by NCI (R01-CA82996) to Heffelfifnger.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Heffelfinger, S., Yan, M., Gear, R. et al. Inhibition of VEGFR2 prevents DMBA-induced mammary tumor formation. Lab Invest 84, 989–998 (2004). https://doi.org/10.1038/labinvest.3700128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700128
