
Abstract
DCL1, the core component for miRNA biogenesis, is itself regulated by miR162 in Arabidopsis. MiRNA-mediated feedback regulation of AtDCL1 is important to maintain the proper level of DCL1 transcripts. However, it is unknown whether the miRNA-mediated regulation of DCL1 is conserved among plants. We analyzed the SmDCL gene family in Salvia miltiorrhiza, an emerging model plant for Traditional Chinese Medicine (TCM) studies, using a comprehensive approach integrating genome-wide prediction, molecular cloning, gene expression profiling and posttranscriptional regulation analysis. A total of five SmDCLs were identified. Comparative analysis of SmDCLs and AtDCLs showed an apparent enlargement of SmDCL introns in S. miltiorrhiza. The absence of miR162 in S. miltiorrhiza and the loss of miR162 target site in SmDCL1 were unexpectedly found. Further analysis showed that the miR162 target site was not present in DCL1 from ancient plants and was gained during plant evolution. The gained miR162 target site might be lost in a few modern plants through nucleotide mutations. Our results provide evidence for the gain and loss of miR162 and its target sites in Dicer-like genes during evolution. The data is useful for understanding the evolution of miRNA-mediated feedback regulation of DCLs in plants.
Similar content being viewed by others


Introduction
Small RNAs are noncoding RNAs of about 20−24 nucleotides in length. They play vital roles in multiple developmental and physiological processes in various organisms through sequence-specific regulation of target genes at the transcriptional or post-transcriptional level1. Based on the biogenesis pathways, plant small RNAs can be classified into two major classes, microRNAs (miRNAs) and small interfering RNAs (siRNAs). SiRNAs are a large small RNA class with four subclasses, including heterochromatic siRNAs (hc-siRNAs), trans-acting siRNAs (ta-siRNAs), natural antisense transcript-derived siRNAs (nat-siRNAs) and long siRNAs (lsiRNAs)2. MiRNAs are produced from transcripts with internal stem-loop structures3, whereas plant siRNAs are derived from inverted repeat sequences, dsRNAs copied from single-stranded RNAs (ssRNA), over-lapping regions of bidirectional transcripts, or dsRNAs formed by virus replication4. Plant small RNAs regulate gene expression by loading into RNA-induced silencing complexes (RISCs) and then interacting with homologous RNA or DNA molecules for direct RNA cleavage, translational repression, or DNA methylation. The biogenesis and function of plant small RNAs involves various families of proteins, such as Dicer-likes (DCLs), HYPONASTIC LEAVES1 (HYL1), C2H2 Zn-finger protein SERRATE (SE), HEN1, HASTY, RNA dependent RNA polymerases (RDRs) and Argonautes (AGOs), of which DCLs are the core components for small RNA biogenesis5,6.
DCLs are multidomain ribonucleases characterized by six domains, including DExD-helicase (DExDc), helicase-C (HELICc), Duf283, PAZ, RIBOc and double stranded RNA-binding (dsRB) domain7. DExDc and HELICc existing in the N- and the C-terminals of the helicase region, respectively, are involved in ATP-dependent RNA or DNA unwinding. The ATP-binding site locates in DExDc domain. PAZ binds single-stranded RNAs with the two-base 3′-overhangs8. The RIBOc domain, known as ribonuclease III C terminal domain, is involved in dsRNA cleavage9, whereas dsRB mediates the discrimination of different RNA substrates and subsequent incorporation of effector complexes7. The function of DUF283 is currently unknown.
DCLs are usually encoded by a multiple gene family in plants. The number of DCL genes in each plant species may be varied. For instance, there are four in Arabidopsis10, five in poplar, maize and sorghum11, seven in tomato12 and eight in rice13. Among them, Arabidopsis DCLs (AtDCLs) are well-studied. Each of the four AtDCLs is primarily associated with the biogenesis of specific small RNA species, but they may play redundant and hierarchical roles in the production of various sRNAs14. AtDCL1 is a core component for miRNA biogenesis, whereas AtDCL2, AtDCL3 and AtDCL4 are mainly involved in the derivation of siRNAs15. AtDCL2 generates 22 nt siRNAs from endogenous inverted-repeats, integrated viruses and transgenes and plays significant roles in virus resistance and transitive silencing of transgenes14,16. AtDCL3 is responsible for the derivation of heterochromatic siRNAs mostly from repetitive DNA loci. These siRNAs are about 24 nt in length and mediate the establishment and maintenance of heterochromatin states through RNA-dependent DNA methylation and histone modification17. AtDCL4 functions in the biogenesis of 21 nt phased siRNAs and ta-siRNAs18. It is also involved in dicing integrated viruses or transgenes into 21 nt siRNAs, which initiate transgene silencing and virus resistance16. These primary siRNAs may further initiate secondary siRNA production under the action of AtDCL2, AtDCL4 and other genes16,19. In addition, the functions of various rice OsDCL genes have been analyzed. OsDCL1 is involved in miRNA biogenesis as its Arabidopsis homolog, AtDCL120. OsDCL4, the homolog of AtDCL4, is responsible for the biogenesis of 21 nt siRNAs associated with inverted repeat transgenes, ta-siRNAs and other 21 nt phased siRNAs and has been found to play a broader role in rice development than AtDCL4 in Arabidopsis21,22. OsDCL3b, rather than OsDCL3a, is involved in the processing of 24 nt phased siRNAs22. The function of DCLs in other plants is poorly understood.
It has been shown that Arabidopsis AtDCL1 and Physcomitrella patens PpDCL1 are negatively regulated by miRNAs. In Arabidopsis, miR162 target AtDCL1 mRNA for direct cleavage at a complementary site formed by the splicing of exon 12 to exon 1323. Additionally, intron 14 of the AtDCL1 primary transcript may form a hairpin structure generating Arabidopsis miR83824. Excision of MIR838 precursor leads to the production of truncated, non-functional AtDCL1 transcripts. It provides a regulatory feedback mechanism supplementing miR162-directed regulation to maintain the proper level of AtDCL1 mRNA24. Similarly, intron 7 of the PpDCL1 primary transcript forms a hairpin structure generating P. patens miR104725. Although miR1047 and miR838 are different in sequence, generate from distinct intron number and arise in an evolutionarily independent manner, miR1047 may play an analogous role of miR838 in the negative feedback regulation of DCL1 in P. patens25. MiRNA-mediated negative feedback loops in other plant DCLs remain to be elucidated.
Salvia miltiorrhiza, which has been widely used for treating dysmenorrhoea, amenorrhoea and cardiovascular disease in China for thousands of years, is not only one of the best selling traditional Chinese medicine (TCM) but also an emerging model plant for TCM studies26,27,28,29,30,31,32,33 With the aim to elucidate the core components of gene silencing pathways in S. miltiorrhiza, we had previously identified the SmAGO and the SmRDR gene families30,34. Here we report the characterization of the SmDCL gene family using a comprehensive approach integrating genome-wide prediction, molecular cloning, gene expression profiling and posttranscriptional regulation analysis. We showed the loss of miR162 target site in SmDCL1 from S. miltiorrhiza. The results shed lights on the regulation and biological functions of SmDCLs.
Results
Identification and molecular cloning of five DCL genes in S. miltiorrhiza
Blast analysis of Arabidopsis and rice DCL amino acid sequences against the current assembly of the S. miltiorrhiza genome31 revealed five SmDCL loci (Fig. 1 and Table 1). Gene models were further predicted for 5 SmDCLs using Genscan (http://genes.mit.edu/GENSCAN.html)35 and corrected manually by comparison with DCL genes identified from other plant species using the BLASTx algorithm (http://www.ncbi.nlm.nih.gov/BLAST)36. The predicted SmDCL cDNAs encode proteins containing DExDc, HELICc, PAZ, dsRB and RNase III domains (Table 2), which are conserved in other plant DCLs and show high sequence similarity with known plant DCLs, such as AtDCLs and OsDCLs, at both the nucleotide and amino acid levels. In order to validate the prediction of SmDCL genes, we cloned and sequenced the 5′ and 3′ ends of SmDCL cDNAs using RNA ligase-mediated rapid amplification of 5′ (5′ RACE) and 3′ (3′ RACE) cDNA ends, respectively. Based on sequence of the cloned 5′ and 3′ ends and the predicted SmDCL cDNAs, we designed primers and then PCR-amplified and sequenced full-length SmDCL cDNAs. Comparison of the cloned SmDCLs and the predicted ones showed that the gene models of SmDCLs were correctly predicted, although a few single nucleotide discrepancies most probably caused by polymorphisms and RT-PCR errors were found between the cloned and the predicted sequences. The results provide five experimentally validated full-length cDNAs of SmDCLs for further systematic characterization. Based on the similarities between SmDCLs and AtDCLs, the five SmDCLs were named SmDCL1, SmDCL2, SmDCL3, SmDCL4a and SmDCL4b, respectively. The cloned SmDCL cDNAs have been deposited in GenBank under accession numbers shown in Table 1.

Gene structures of DCLs in S. miltiorrhiza and Arabidopsis.
Exons are indicated in green boxes. UTRs are shown in blue boxes. Introns are indicated in lines.
Comparative analysis of SmDCLs and AtDCLs in sequence features, gene structures and conserved domains
Analysis of the cloned SmDCL cDNA showed that the length of open reading frames (ORFs) of SmDCLs varied between 4,158 (SmDCL2) and 5,772 bp (SmDCL1), 5′ untranslated regions (UTRs) varied from 45 (SmDCL4a) to 328 bp (SmDCL1), while 3′ UTRs varied between 166 (SmDCL4a) and 454 bp (SmDCL4b) (Table 1). The size of deduced SmDCL proteins varies between 1385 (SmDCL2) and 1927 (SmDCL1) amino acids, the molecular weight (Mw) varies from 156.3 (SmDCL2) to 216.4 kDa (SmDCL1) and the theoretical pI varies between 6.01 (SmDCL1) and 7.10 (SmDCL2) (Table 1). These sequence features of SmDCLs are quite similar to those of AtDCLs (Table 1). For instance, all SmDCLs and AtDCLs have the theoretical pI of about 6–7. SmDCL1 and AtDCL1 are the largest among DCL proteins in S. miltiorrhiza and A. thaliana, respectively. Additionally, the overall size of SmDCL proteins is comparable with the corresponding AtDCLs (Table 1).
Alignment of the cloned SmDCL cDNA with the corresponding genomic sequence showed that the intron number of SmDCLs varied from 19 (SmDCL1) to 24 (SmDCL3) (Table 1). SmDCL1 and AtDCL1 contain 19 introns and have very similar exon patterns (Fig. 1). The similarity of intron number and exon patterns was also found for other DCL gene pairs from S. miltiorrhiza and A. thaliana (Fig. 1). It suggests the conservation of DCLs in S. miltiorrhiza and A. thaliana. Interestingly, we observed an apparent enlargement of DCL introns in S. miltiorrhiza compared with Arabidopsis (Fig. 1). The expansion of intron size is probably due to the proliferation of transposable elements (TEs) during evolution or domestication of S. miltiorrhiza. However, it is necessary to further investigate the characteristics of introns in SmDCLs for elucidating the actual mechanism of intron size expansion.
Multiple sequence alignment of the deduced amino acid sequences using T-Coffee37 showed various conserved regions among SmDCLs (see Supplementary Fig. S1 online). Search of the deduced SmDCL proteins for conserved domains against the NCBI Conserved Domain Database (CCD) revealed that SmDCLs contained DExDc, HELICc, PAZ, dsRB and RIBOc domains (Table 2). These domains located in the conserved regions identified using T-Coffee37 and were also found in animal, fungal and other plant DCL proteins, suggesting the conservation of DCLs in organisms.
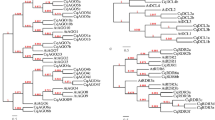
Phylogenetic tree construction for DCL proteins in S. miltiorrhiza, Arabidopsis and rice
An unrooted neighbor-joining (NJ) tree was constructed for determining the relationship of five SmDCLs, four AtDCLs and eight OsDCLs using MEGA4.038 (Fig. 2). Based on the NJ tree, the referred DCL proteins fall into four clades. SmDCL1 clusters with AtDCL1, OsDCL1a, OsDCL1b and OsDCL1c in the DCL1 clade. SmDCL2 is closely related to AtDCL2, OsDCL2a and OsDCL2b in the DCL2 clade. SmDCL3, AtDCL3, OsDCL3a and OsDCL3b belong to the DCL3 clade. SmDCL4a and SmDCL4b cluster with AtDCL4 and OsDCL4 in the DCL4 clade. It suggests that each of four DCL clades include at least a SmDCL, implying the deeply conserved roles of SmDCLs with their counterparts in Arabidopsis and rice. Interestingly, two SmDCLs, including SmDCL4a and SmDCL4b, were found in the DCL4 clade. To our best knowledge, it is the first time to find two DCLs in a plant belonging to the DCL4 clade. SmDCL4a and SmDCL4b show similar exon patterns, whereas the size of various SmDCL4a and SmDCL4b intron is distinct (Fig. 1). For instance, introns 4, 5, 7, 15, 16 and 20 of SmDCL4a are longer than the corresponding introns in SmDCL4b, while introns 2, 8, 11 and 17 of SmDCL4a are apparently shorter (Fig. 1). It indicates that intron size expansion and condensation happened in SmDCL4a and SmDCL4b.

Expression patterns of SmDCL genes in S. miltiorrhiza
As the core components for small RNA biogenesis, DCLs play vital roles in plant development5,6. The expression pattern of DCLs may be correlated with their physiological functions. With the aim to primarily elucidate the functions of SmDCLs, we analyzed the expression level of SmDCL genes in flowers, leaves, stems and roots of 2-year-old, field nursery-grown S. miltiorrhiza using quantitative RT-PCR technology. The results showed that all of five SmDCLs were expressed in S. miltiorrhiza tissues analyzed, although differential expression patterns were observed (Fig. 3). It is consistent with the significant role of DCLs in the biogenesis of miRNAs and siRNAs involving in plant development and stress responses. SmDCL1 showed the highest expression in flowers, followed by roots and leaves and less in stems (Fig. 3a). The pattern is very similar with that of its Arabidopsis counterpart, AtDCL1, which is consistent with the conserved roles of SmDCL1 and AtDCL1 in miRNA biogenesis. The expression pattern of SmDCL2 is similar with that of SmDCL4a showing more root-specific (Fig. 3b,d). Consistently, all of their Arabidopsis counterparts, AtDCL2 and AtDCL4, are involved in transgene silencing and virus resistance, although AtDCL2 generates 22 nt siRNAs, while AtDCL4 functions in the biogenesis of 21 nt siRNAs16. It is noticed that SmDCL4b show a distinct expression pattern with SmDCL2 and SmDCL4a (Fig. 3e), although SmDCL4a and SmDCL4b cluster in a clade (Fig. 2) and have similar exon patterns (Fig. 1). It indicates that SmDCL4a and SmDCL4b may play different roles in the production of siRNAs. Functional divergence of DCLs within a clade was previously found for rice OsDCL3a and OsDCL3b22. Both OsDCL3a and OsDCL3b belong to the DCL3 clade, whereas the processing of 24 nt phased small RNAs requires OsDCL3b rather than OsDCL3a in rice22. In S. miltiorrhiza, only one SmDCL3 was identified. It expressed in flowers, leaves, stems and roots of S. miltiorrhiza at the similar levels (Fig. 3c), which seems to be consistent with the role of DCLs in the DCL3 clade, such as AtDCL3 and OsDCL3b, in the derivation of heterochromatic siRNAs17.

Expression of SmDCLs in flowers (Fl), leaves (Le), stems (St) and roots (Rt) of S. miltiorrhiza.
Expression levels were quantified by qRT-PCR. The levels in roots were arbitrarily set to 1. Error bars represent the standard deviations of three technical PCR replicates.
Analysis of miRNA-mediated regulation of SmDCLs
Arabidopsis AtDCL1 and P. patens PpDCL1 involved in miRNA biogenesis are themselves regulated by miRNAs23,24,25. AtDCL1 is directly cleaved by miR16223. The level of AtDCL1 mRNA is also affected by the excision of MIR838 precursor from intron 14 of AtDCL1 primary transcripts24. Similarly, the level of PpDCL1 mRNA in P. patens cells is negatively regulated by the generation of miR1047 from intron 7 of PpDCL1 primary transcripts25. In order to know whether there is a miRNA-mediated feedback regulation of SmDCL1, we first analyzed the secondary structure of all 19 introns in SmDCL1. No stem-loop structures meeting the widely used criteria for miRNA precursors were predicted39. We next performed a target search of plant miRNAs in miRBase against SmDCL1 and the other four SmDCLs using psRNATarget40,41. With the maximum expectation of 3.0 applied in the target search, a total of 10 miRNA familes, including miR397, miR1035, miR1536, miR4395, miR4407, miR2873, miR5164, miR5247 and miR5303 were identified. Further alignment of these miRNA sequences with the current assembly of the S. miltiorrhiza genome using SOAP2 with two mismatches allowed42 and secondary structure prediction for genomic DNA fragments surrounding these miRNA sequences using the mfold program43 allowed us to identify a precursor for miR397 (Fig. 4a). No precursors were predicted for the other 9 miRNAs, indicating they could be not present in S. miltiorrhiza. The identified S. miltiorrhiza miR397 showed near-perfect complementarity to SmDCL1 with a penalty score of 3.544 (Fig. 4b). Using the modified 5′-rapid amplification of cDNA ends (RACE) method44, we tested whether SmDCL1 were authentic targets of miR397. Unfortunately, no RACE products were obtained for the predicted cleavage after repeated experiments.

The Sm-MIR397 precursor and complementarities between miRNAs and SmDCL1.
(a) Predicted hairpin structures of Sm-MIR397. Mature miRNA sequences are indicated in red. Vertical lines indicate G:C and A:U pairings. Circles indicate G:U pairings. (b) Complementarities between Sm-miR397, At-miR162a/b and SmDCL1. The heavy black line represents ORF. The lines flanking ORF represent nontranslated regions. MiRNA complementary sites with the nucleotide positions of SmDCL1 cDNA are indicated. The RNA sequence of each complementary site from 5′ to 3′ and the predicted miRNA sequence from 3′ to 5′ are shown in the expanded regions. Arrows indicate the 5′ termini of three cDNA fragments (c) with the frequency of clones (in parentheses) and the nucleotide positions of SmDCL1 cDNA shown. (c) Determination of the 5′ termini of truncated SmDCL1 cDNA fragments using the 5′-RACE method. Nested PCR products were separated in a 2% agarose gel.
Loss of miR162 target site in SmDCL1 and lack of miR162 in S. miltiorrhiza
In Arabidopsis, AtDCL1 is an experimentally validated target of miR16223. However, it was not among the miRNAs predicted to target DCLs for cleavage in S. miltiorrhiza. Manual alignment of miR162 sequence from Arabidopsis with SmDCL1 showed that the penalty scores for mismatched pattern in the miR162:SmDCL1 duplex within a 20-base sequence window was 5.0 (Fig. 4b)44. Analysis of the target site variation between A. thaliana and A. lyrata for the highly conserved miRNA families showed that 10% of the A. thaliana miRNA-target pairs were lost45. In order to know whether the mismatched patterns of miR162:SmDCL1 duplexes were conserved in different S. miltiorrhiza cultivation lines, we cloned SmDCL1 cDNA fragments corresponding to the complementary sites of miR162 from the other two S. miltiorrhiza lines, namely 992 and shh. The result showed that the sequence of the complementary sites of miR162 in lines 992 and shh was identical to that in line 993 (Fig. 5), suggesting the conservation of the mismatched patterns of miR162:SmDCL1 duplex in three lines of S. miltiorrhiza analyzed.

Alignment of the miR162 complementary site in DCL1s from various plant species.
A. thaliana At-miR162a/b is also shown. Watson-Crick pairing is indicated by vertical dashes. Penalty scores for mismatched pattern in the miR162:DCL1 duplex within a 20-base sequence window calculated as described previously are shown in parentheses (Lu et al. 2005). The sequences analyzed include Arabidopsis AtDCL1 (AT1G01040), Arachis hypogaea AhDCL1 (JR564267), Brachypodium distachyon BdDCL1 (XM_003558898), Camelina sativa CamDCL1 (GABO01016802), Cannabis sativa CanDCL1 (JP472773), Catharanthus roseus CrDCL1 (GACD01069741), Chorispora bungeana CbDCL1 (KA047874), Chromolaena odorata CoDCL1 (GACH01012147), Cucumis sativus CsDCL1 (XM_004155222), Elaeocarpus photiniifolius EpDCL1 (FX137492), Fragaria vesca FvDCL1 (XM_004308223), Gerbera hybrid cultivar GerDCL1 (GACN01020550), Glycine max GmDCL1 (XM_003553757), Humulus lupulus HlDCL1 (GAAW01068254), Ipomoea batatas IbDCL1 (JP112449), Lactuca serriola LsDCL1 (JO020520), Medicago truncatula MtDCL1 (XM_003558898), Musa acuminata MaDCL1 (JV351655), Nicotiana benthamiana NbDCL1 (KA746219), Olea europaea OeDCL1 (GABQ01046272), Oncidium ‘Gower Ramsey’ OncDCL1 (JL935168), Oryza sativa OsDCL1a (LOC_Os03g02970), Physalis peruviana PhyDCL1 (JO133983), Physcomitrella patens PpDCL1 (XM_001757896), Populus trichocarpa PtDCL1 (XM_002302643), Rehmannia glutinosa RgDCL1 (JG014336), Ricinus communis RcDCL1 (XM_002515051), Salvia miltiorrhiza line 993 SmDCL1(993) (KF366499), Salvia miltiorrhiza line 992 SmDCL1(992), Salvia miltiorrhiza line ssh SmDCL1(ssh), Saussurea involucrate SauDCL1 (JW888406), Selaginella moellendorffii SelDCL1 (XM_002965595), Sesanum indicum SiDCL1 (JP640291), Silene latifolia SilDCL1 (JO777655), Solanum lycopersicum SlDCL1 (10G005130), Vitis vinifera VvDCL1 (XM_002268333), Zea mays ZmDCL1 (DY397446).
To test whether SmDCL1 is regulated by miR162, the modified 5′-RACE analysis was carried out. After nested and nesting PCR amplification, at least ten cDNA bands were obtained (Fig. 4c). Sequence analysis of three cDNA bands with the approximately expected size showed that the 5′ end of PCR products located at upstream 52 bp, downstream 46 and 81 of the predicted cleavage site, respectively (Fig. 4b), suggesting they were not miR162-directed cleavage products.
Since the loss of miR162 target site in SmDCL1, we ask whether miR162 is present in S. miltiorrhiza. In order to address this question, we checked the published high-throughput sRNA sequencing data for mature miR162 sequence in S. miltiorrhiza46. No miR162 sequence was found in small RNA libraries for S. miltiorrhiza roots, stems and leaves. The read of miR162 sequence in flower small RNA library was only one. Extremely low small RNA reads could be a result from next-generation sequencing contamination47. To test this possibility, we first searched the current assembly of the S. miltiorrhiza genome for miR162 precursors. No positive results were obtained. Next, we searched our S. miltiorrhiza small RNA database for mature miR162 sequence. The database contains 114,426,648 clean reads obtained by high throughput Solexa sequencing of 18–30 nt small RNAs from flowers, leaves, stems and roots of S. miltiorrhiza plants. Consistently, no miR162 sequence was identified. Taken together, it is highly likely that miR162 is absent from S. miltiorrhiza.
Mismatched patterns in the miR162:DCL1 duplexes from 35 plant species
In order to know whether the absence of miR162-mediated feedback regulation of DCL1 is widely present in plants or just limited to S. miltiorrhiza or a few plant species, an examination of the miR162 complementary site in DCL1s from 35 plant species was carried out. The cDNA regions complementary to miR162 are highly conserved among plant DCLs, except SmDCL1, Physcomitrella patens PpDCL1, Selaginella moellendorffii SelDCL1, Rehmannia glutinosa RgDCL1, Sesamum indicum SiDCL1 and Olea europaea OeDCL1 (Fig. 5). It suggests the conservation of miR162-mediated feedback regulation of DCL1 in most plants. PpDCL1 and SelDCL1 with the penalty score for mismatched patterns in the miR162:DCL1 duplexes to be 9.0 and 7.0, respectively (Fig. 5), have been confirmed to be not regulated by miR16225. The penalty score for miR162:RgDCL1, miR162:SiDCL1 and miR162:OeDCL1 duplexes is 3.0 (Fig. 5). No miR162 was found in more than 13 million unique sequences obtained by high throughput Solexa sequencing of 18–20 nt small RNAs from leaves, stems and roots of the first and second year cropping R. glutinosa plants48. Similarly, no miR162 was found in about 94 million sequence reads from juvenile and adult shoots, ripe and unripe fruits and leaves of O. europaea49,50. It indicates that the miR162-mediated feedback regulation of DCL1 seemed to be absent from R. glutinosa and O. europaea. The regulation of SiDCL1 remains to be elucidated.
Discussion
Although DCLs have been identified from various plant species, functional characterization of DCLs is limited to a few plants, such as Arabidopsis and rice18,19,20. The identification and molecular cloning of five SmDCLs provides a base for elucidating the function of SmDCLs and for understanding the biogenesis pathways and functions of small RNAs in S. miltiorrhiza, an emerging model plant with high medicinal value26. Five SmDCLs cluster into four clades with Arabidopsis and rice DCLs (see Supplementary Fig. S1 online), indicating the existence of four types of DCLs with distinct functions in S. miltiorrhiza as the cases in Arabidopsis and rice7,13,51. Conservation of sequence features, gene structures and functional domains implies that the function of each SmDCL could be similar to its Arabidopsis and rice counterparts in the same clade. However, it is interesting to show, for the first time, two SmDCLs in the DCL4 clade. SmDCL4a and SmDCL4b have similar exon patterns, but the intron size is distinct with some intron expanded while the others condensed (Fig. 1). Moreover, SmDCL4a and SmDCL4b showed distinct expression patterns (Fig. 3). These results indicate that SmDCL4a and SmDCL4b may play different roles in S. miltiorrhiza as the case of OsDCL3a and OsDCL3b in rice22. Further production and analysis of transgenic S. miltiorrhiza plants with SmDCL4a and/or SmDCL4b up- or down-regulated will definitely shed light on the biological function of SmDCL4a and SmDCL4b.
It has been shown the presence of miRNA-mediated feedback regulation of Arabidopsis AtDCL1 and P. patens PpDCL123,24,25. AtDCL1 is regulated by miR162 and miR83823,24,25, while PpDCL1 is regulated by miR104725. Analysis of the regulation mechanism of SmDCLs unexpectedly revealed the loss of miR162 target site in SmDCL1. Close examination of the miR162 complementary regions showed the absence of miR162 target sites in DCL1 from the non-vascular plant P. patens and the ancient vascular plant S. moellendorffii25,52, suggesting that the miR162 target site was not present in ancient plants and was gained during plant evolution. On the other hand, the gained miR162 target site might be lost in a few modern plants, such as S. miltiorrhiza. Since S. miltiorrhiza is evolutionarily far from P. patens and S. moellendorffii compared with many plants with the conserved miR162 target site (Fig. 5), gain and loss of miR162 target sites seems to be two independent events during plant evolution. Gain and loss of miRNA target sites has been previously investigated in Arabidopsis and rice45,53. The loss of miRNA target sites was proposed to be a consequence of gene ortholog loss, target site sequence disruption, or point substitutions/nucleotide mutations45,53. Analysis of the miR162 target sites (except the bulge nucleotide) showed single nucleotide mutation in S. indicum SiDCL1 and O. europaea OeDCL1, two in R. glutinosa RgDCL1, while four in S. miltiorrhiza SmDCL1 (Fig. 5). It suggests the loss of miR162 target sites was caused by nucleotide mutations rather than gene ortholog loss and target site sequence disruption.
It has been generally considered that miRNAs and their targets co-evolve in animals54. The absence of miR162 target site goes along with the lack of miR162 in P. patens52, S. moellendorffii25, R. glutinosa46, O. europaea49,50 and S. miltiorrhiza, suggesting that the miR162 gene, similar to the miR162 target site, might be lost in some modern plants during plant evolution and indicating the possibility for co-evolution of miR162 and miR162 target sites in plants. However, since current information is preliminary, it is impossible to make a conclusion. Relatively frequent gain and loss of miRNA genes has been previously reported in A. thaliana55. Analysis of miRNA-target pair conservation between A. thaliana and A. lyrata showed that about 12.5% of non-conserved pairs were due to the loss of corresponding miRNAs in A. lyrata45. Of the 387 miRNAs from wild rice, 259 were not found in cultivated rice, suggesting a significant loss of miRNAs during rice domestication56. A possible mechanism for miRNA gene loss is nucleotide mutation. For instance, among 591 rice miRNAs, 364 have one or more SNPs in their precursor sequences57. SNPs in the stem regions may cause unstable of the miRNA hairpin structures, while SNPs in mature miRNAs have great potential to loss miRNA-target interaction56. Genome-wide duplication could be the other possible mechanism for the loss of miRNA genes. Comparative analysis of miRNA genes in maize and sorghum showed that duplicated miRNA genes underwent extensive gene-loss, with about 35% of ancestral sites were retained as duplicate homoeologous miRNA genes58. Since there is no information for miR162 gene variation among S. miltiorrhiza and its relative species and it is unknown for the genome-wide duplication events happened during S. miltiorrhiza evolution, the mechanism for loss of miR162 in S. miltiorrhiza is currently unknown and need to be further investigated.
It has been proposed that miR162-mediated feedback regulation of DCL1 is important in maintaining AtDCL1 at functionally sufficient, but not limiting or excessive, levels23 and the excision of MIR838 precursor from AtDCL1 primary transcript, which leads to the production of truncated and non-functional AtDCL1 transcripts, provides a regulatory feedback mechanism supplementing miR162-directed regulation to maintain the proper level of AtDCL1 mRNA24. Additionally, P. patens miR1047 seems to play a similar role in feedback regulation of PpDCL125. However, data for the actual physiological functions of miR162, miR838 and miR1047 is lacking. Without direct physiological evidence, the significance of miRNA-mediated feedback regulation of DCL1 is largely uncertained. The absence of miR162-mediated feedback regulation of DCL1 in S. miltiorrhiza and probably in R. glutinosa and O. europaea implies that, at least in some plant species, miR162-mediated feedback mechanism could be not vital. It is possible that an alternative mechanism for maintaining SmDCL1 at a proper level exists in S. miltiorrhiza and other plant species lacking the miR162-mediated feedback regulation of DCL1. Further investigating the regulatory mechanism of SmDCLs using transgenics may help to demonstrate the significance of miRNA-mediated feedback regulation of DCL1 in plants and reveal the alternative of this feedback regulation in S. miltiorrhiza.
Methods
Plant materials
S. miltiorrhiza Bunge (line 993) was cultivated under natural growth conditions in a field nursery located at the Institute of Medicinal Plant Development, Beijing, China. Mature flower buds, mature and healthy leaves, young stems and roots in about 0.5 cm diameter were collected from two-year-old plants on August 15th, 2012. Tissues were collected from at least 3 plants and then pooled. The pooled tissues were stored in liquid nitrogen until use.
Prediction and cloning of SmDCL genes
SmDCL genes were identified by tBLASTn analysis36 of Arabidopsis and rice DCL protein sequences (http://www.ncbi.nlm.nih.gov/protein) against the current assembly of the S. miltiorrhiza genome31. All retrieved DNA sequences were used for gene prediction on the Genscan web server (http://genes.mit.edu/GENSCAN.html)35. The predicted gene models were further examined and corrected manually by comparison with DCL genes identified from other plant species using the BLASTx algorithm (http://www.ncbi.nlm.nih.gov/BLAST)36.
To clone the full-length SmDCL cDNAs, RNA ligase-mediated rapid amplification of 5′ cDNA ends (5′-RACE) and 3′ cDNA ends (3′-RACE) was carried out using the GeneRacer kit (Invitrogen, Carlsbad, CA, USA). PCR amplification was performed using the following conditions: pre-denaturation at 94 °C for 2 min, 5 cycles of amplification at 94 °C for 30 s and 72 °C for 1 min, 5 cycles of amplification at 94 °C for 30 s and 70 °C for 1 min, 25 cycles of amplification at 94 °C for 30 s, 56 °C for 30 s and 72 °C for 2 min, followed by a final extension at 72 °C for 15 min. Nested PCR amplifications were carried out using the following conditions: pre-denaturation at 94 °C for 2 min, 30 cycles of amplification at 94 °C for 30 s, 58 °C for 30 s and 72 °C for 2 min, followed by a final extension at 72 °C for 15 min. PCR products were gel-purified, cloned and sequenced. The nesting and nested gene-specific primers used for 5′- and 3′-RACE are listed in Supplementary Table S1 and S2 online, respectively. Full-length SmDCL cDNAs were amplified using gene-specific forward and reverse primers (see Supplementary Table S3 online) under the following conditions: pre-denaturation at 94 °C for 2 min, 30 cycles of amplification at 94 °C for 30 s, 56 °C for 30 s and 72 °C for 3 min, followed by a final extension at 72 °C for 15 min. PCR products were gel-purified and cloned. For each transformation, three clones were sequenced at Beijing Sunbiotech Co., Ltd (Beijing, China). Sequences from three clones were aligned with the predicted SmDCL sequence using DNAMAN (Lynnon BioSoft, San Ramon, CA, USA). The cloned cDNAs showing the least nucleotide discrepancies with the predicted sequences were selected and deposited in GenBank (Table 1).
Phylogenetic tree construction and bioinformatics analysis
Phylogenetic tree was constructed using MEGA version 4.0 by the neighbor-joining method with 1000 bootstrap replicates38,59. Intron/exon structures were analyzed manually based on genomic DNA sequences and the cloned cDNA sequences. Molecular weight (MW) and theoretical isoelectric point (pI) were predicted using DNAMAN. Conserved domains were analyzed by search the deduced amino acid sequence of SmDCLs against the NCBI conserved domain (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Multiple sequence alignment of the deduced SmDCL amino acid sequences was carried out using T-Coffee37.
Quantitative real-time reverse transcription-PCR (qRT-PCR)
Total RNA was isolated from plant tissues using the plant total RNA extraction kit (BioTeke, Beijing, China) and genomic DNA was removed by treating with RNase-free DNase (Promega, Madison, WI, USA). One μg total RNA was converted into cDNA by 200 U Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA) in a 20 μl volume. cDNA was diluted into 200 μl and then used for qRT-PCR. Gene-specific primers were listed in Supplementary Table S4 online. SmUBQ10 was used as a control as previously described28. PCR was carried out in a 20 μl volume containing 2 μl diluted cDNA, 250 nM forward primer, 250 nM reverse primer and 1 × SYBR Premix Ex Taq II (TaKaRa Bio, Otsu, Japan) using the following conditions: pre-denaturation at 95 °C for 30 s, 40 cycles of amplification at 95 °C for 5 s, 60 °C for 18 s and 72 °C for 15 s. The results from gene-specific amplification were analyzed using the comparative Cq method, which uses an arithmetic formula, 2-ΔΔCq, to achieve results for relative quantification60. Cq represents the threshold cycle.
Identification of S. miltiorrhiza miRNAs with perfect or near-perfect complementarity to SmDCLs
Plant miRNAs with the potential to target SmDCLs for cleavage were predicted using psRNATarget with the default parameters40. Known plant miRNAs were downloaded from miRBase (release 19, http://www.mirbase.org/)41. The identified miRNAs were then aligned with the current assembly of the S. miltiorrhiza genome31 using SOAP2 with no more than 2 mismatches allowed42. S. miltiorrhiza genomic DNA sequences with known plant miRNAs aligned were predicted for hairpin structures using mfold43. Criteria described by Meyers et al39 were applied to annotate S. miltiorrhiza miRNAs.
5′RLM-RACE for analysis of miRNA-directed cleavage of SmDCLs
The modified RNA ligase-mediated rapid amplification of 5′ cDNAs method (5′RLM-RACE) was performed using the GeneRacer kit (Invitrogen, Carlsbad, CA, USA) as described previously44. PCRs were carried out on mRNA isolated from pooled S. miltiorrhiza tissues containing flowers, leaves, stem and roots. Gene-specific primers used in this experiment are listed in Supplementary Table S5 online.
PCR amplification of SmDCL1 cDNA fragments in S. miltiorrhiza lines 992 and shh
SmDCL1 cDNA fragments surrounding the predicted miR162 target site were PCR-amplified on cDNA from the leaves of S. miltiorrhiza lines 992 and shh using 5′-GTCAGGGAGGAGCTGTGACAATT-3′ as the forward primer and 5′-CGTACATGAAAGCTCTTGAGCGAT-3′ as the reverse primer.
Additional Information
How to cite this article: Shao, F. et al. Comparative analysis of the Dicer-like gene family reveals loss of miR162 target site in SmDCL1 from Salvia miltiorrhiza. Sci. Rep. 5, 9891; doi: 10.1038/srep09891 (2015).
References
Kidner, C. A. & Martienssen, R. A. The developmental role of microRNA in plants. Curr. Opin. Plant Biol. 8, 38–44 (2005).
Katiyar-Agarwal, S., Gao, S., Vivian-Smith, A. & Jin, H. A novel class of bacteria-induced small RNAs in Arabidopsis. Genes Dev. 21, 3123–3134 (2007).
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism and function. Cell 116, 281–297 (2004).
Zamore, P. D., Tuschl, T., Sharp, P. A. & Bartel, D. P. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101, 25–33 (2000).
Fang, Y. & Spector, D. L. Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr. Biol. 17, 818–823 (2007).
Song, L., Han, M. H., Lesicka, J. & Fedoroff, N. Arabidopsis primary microRNA processing proteins HYL1 and DCL1 define a nuclear body distinct from the Cajal body. Proc. Natl. Acad. Sci. U. S. A. 104, 5437–5442 (2007).
Margis, R. et al. The evolution and diversification of Dicers in plants. FEBS Lett 580, 2442–2450 (2006).
Kini, H. K. & Walton, S. P. In vitro binding of single-stranded RNA by human Dicer. FEBS Lett. 581, 5611–5616 (2007).
Tahbaz, N. et al. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep. 5, 189–194 (2004).
Finnegan, E. J. & Matzke, M. A. The small RNA world. J. Cell Sci. 116, 4689–4693 (2003).
Qian, Y. et al. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 30, 1347–1363 (2011).
Bai, M. et al. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene 501, 52–62 (2012).
Kapoor, M. et al. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics 9, 451 (2008).
Deleris, A. et al. Hierarchical action and inhibition of plant Dicer-like proteins in antiviral defense. Science 313, 68–71 (2006).
Bouché, N., Lauressergues, D., Gasciolli, V. & Vaucheret, H. An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J. 25, 3347–3356 (2006).
Mlotshwa, S. et al. DICER-LIKE2 plays a primary role in transitive silencing of transgenes in Arabidopsis. PLoS ONE 3, e1755 (2008).
Xie, Z. et al. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2, e104 (2004).
Xie, Z., Allen, E., Wilken, A. & Carrington, J. C. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 102, 12984–12989 (2005).
Wang, X. B. et al. The 21-nucleotide, but not 22-nucleotide, viral secondary small interfering RNAs direct potent antiviral defense by two cooperative argonautes in Arabidopsis thaliana. Plant Cell 23, 1625–1638 (2011).
Liu, B. et al. Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol 139, 296–305 (2005).
Liu, B. et al. Oryza sativa dicer-like4 reveals a key role for small interfering RNA silencing in plant development. Plant Cell 19, 2705–2718 (2007).
Song, X. et al. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 69, 462–474 (2012).
Xie, Z., Kasschau, K. D. & Carrington, J. C. Negative feedback regulation of Dicer-Like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr. Biol. 13, 784–789 (2003).
Rajagopalan, R., Vaucheret, H., Trejo, J. & Bartel, D. P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 20, 3407–3425 (2006).
Axtell, M. J., Snyder, J. A. & Bartel, D. P. Common functions for diverse small RNAs of land plants. Plant Cell 19, 1750–1769 (2007).
Cheng, T. O. Danshen a popular chinese cardiac herbal drug. J. Am. Coll. Cardiol. 47, 1487–1501 (2006).
He, S. et al. Compound astragalus and Salvia miltiorrhiza extract inhibits cell proliferation, invasion and collagen synthesis in keloid fibroblasts by mediating TGF-beta/smad pathway. Br. J. Dermatol 166, 564–574 (2012).
Ma, Y. et al. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J. Exp. Bot. 63, 2809–2823 (2012).
Hou, X., Shao, F., Ma, Y. & Lu, S. The phenylalanine ammonia-lyase gene family in Salvia miltiorrhiza: genome-wide characterization, molecular cloning and expression analysis. Mol. Biol. Rep. 40, 4301–4310 (2013).
Shao, F. & Lu, S. Genome-wide identification, molecular cloning, expression profiling and posttranscriptional regulation analysis of the Argonaute gene family in Salvia miltiorrhiza, an emerging model medicinal plant. BMC Genomics 14, 512 (2013).
Song, J. et al. Salvia miltiorrhiza as medicinal model plant. Yao Xue Xue Bao 48, 1099−1106 (2013).
Zhang, L. et al. Genome-wide analysis and molecular dissection of the SPL gene family in Salvia miltiorrhiza. J. Integr. Plant Biol. 56, 38–50 (2014).
Li, C. & Lu, S. Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genomics 15, 277 (2014).
Shao, F. & Lu, S. Identification, molecular cloning and expression analysis of five RNA-dependent RNA polymerase genes in Salvia miltiorrhiza. PloS ONE 9, e95117 (2014).
Burge, C. B. & Karlin, S. Finding the genes in genomic DNA. Curr. Opin. Struct. Biol. 8, 346–354 (1998).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 (2000).
Kumar, S., Tamura, K. & Nei, M. MEGA3 integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 5, 150–163 (2004).
Meyers, B. C. et al. Criteria for annotation of plant microRNAs. Plant Cell 20, 3186–3190 (2008).
Dai, X., Zhuang, Z. & Zhao, P. X. Computational analysis of miRNA targets in plants: current status and challenges. Brief Bioinform. 12, 115–121 (2011).
Kozomara, A. & Griffiths-Jones, S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, 152–157 (2010).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Lu, S. et al. Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 17, 2186–2203 (2005).
Fahlgren, N. et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22, 1074–1089 (2010).
Xu, X. et al. Deep sequencing identifies tissue-specific microRNAs and their target genes involving in the biosynthesis of tanshinones in Salvia miltiorrhiza. PLoS ONE 6, e111679 (2014).
Tosar, J. P., Rovira, C., Naya, H. & Cayota, A. Mining of public sequencing databases supports a non-dietary origin for putative foreign miRNAs: underestimated effects of contamination in NGS. RNA 20, 754–757 (2014).
Yang, Y. et al. Differential miRNA expression in Rehmannia glutinosa plants subjected to continuous cropping. BMC Plant Biol. 11, 53 (2011).
Donaire, L., Pedrola, L., Rosa Rde, L. & Llave, C. High-throughput sequencing of RNA silencing-associated small RNAs in olive (Olea europaea L.). PLoS ONE 6, e27916 (2011).
Yanik, H. et al. Genome-wide identification of alternate bearing-associated microRNAs (miRNAs) in olive (Olea europaea L.). BMC Plant Biol. 13, 10 (2013).
Henderson, I. R. et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 38, 721–725 (2006).
Arazi, T. et al. Cloning and characterization of micro-RNAs from moss. Plant J 43, 837–848 (2005).
Guo, X. et al. Selection and mutation on microRNA target sequences during rice evolution. BMC Genomics 9, 454 (2008).
Tang, T. et al. Adverse interactions between micro-RNAs and target genes from different species. Proc. Natl. Acad. Sci. U. S. A. 107, 12935–12940 (2010).
Fahlgren, N. et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE 2, e219 (2007).
Wang, Y. et al. Genomic dissection of small RNAs in wild rice (Oryza rufipogon): lessons for rice domestication. New Phytol. 196, 914–925 (2012).
Liu, Q., Wang, H., Zhu, L., Hu, H. & Sun, Y. Genome-wide identification and analysis of miRNA-related single nucleotide polymorphisms (SNPs) in rice. Rice 6, 10 (2013).
Zhang, L. et al. A genome-wide characterization of microRNA genes in maize. PLoS Genetics 5, e1000716 (2009).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Acknowledgements
We thank Dr. Shilin Chen and the S. miltiorrhiza sequencing group for kindly providing the S. miltiorrhiza genome sequence. We appreciate Prof. Xian’en Li for providing S. miltiorrhiza plants. This work was supported by grants from the Natural Science Foundation of China (grant no. 31370327), the Beijing Natural Science Foundation (grant nos. 5112026 and 5152021), the Major Scientific and Technological Special Project for Significant New Drugs Creation (grant no. 2012ZX09301002-001-031), the Research Fund for the Doctoral Program of Higher Education of China (20111106110033), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT, grant no. IRT1150) and the Fund for Postgraduate Innovation in Peking Union Medical College (2012-1007-026).
Author information
Authors and Affiliations
Contributions
F.S. analyzed the data, performed qRT-PCR and RACE and participated in writing the manuscript. D.Q. assisted in interpreting the experiments. S.L. designed the experiment, analyzed the data and wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shao, F., Qiu, D. & Lu, S. Comparative analysis of the Dicer-like gene family reveals loss of miR162 target site in SmDCL1 from Salvia miltiorrhiza. Sci Rep 5, 9891 (2015). https://doi.org/10.1038/srep09891
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09891
This article is cited by
-
Identification and characterization of Dicer-like genes in leaf rust pathogen (Puccinia triticina) of wheat
Functional & Integrative Genomics (2020)
-
Systematic analysis of DEMETER-like DNA glycosylase genes shows lineage-specific Smi-miR7972 involved in SmDML1 regulation in Salvia miltiorrhiza
Scientific Reports (2018)
-
Characterization of the polyphenol oxidase gene family reveals a novel microRNA involved in posttranscriptional regulation of PPOs in Salvia miltiorrhiza
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
