
Abstract
In this study, 111 Cryptosporidium parvum IId isolates from several species of animals in China, Sweden and Egypt were subtyped by multilocus sequence typing (MLST). One to eleven subtypes were detected at each of the 12 microsatellite, minisatellite and single nucleotide polymorphism (SNP) loci, forming 25 MLST subtypes. Host-adaptation and significant geographical segregation were both observed in the MLST subtypes. A clonal population structure was seen in C. parvum IId isolates from China and Sweden. Three ancestral lineages and the same RPGR sequence were shared by these isolates examined. Therefore, the present genetic observations including the higher nucleotide diversity of C. parvum IId GP60 sequences in Western Asia, as well as the unique distribution of IId subtypes (almost exclusively found in Asia, Europe and Egypt) and in combination with the domestication history of cattle, sheep and goats, indicated that C. parvum IId subtypes were probably dispersed from Western Asia to other geographical regions. More population genetic structure studies involving various C. parvum subtype families using high-resolution tools are needed to better elucidate the origin and dissemination of C. parvum in the world.
Similar content being viewed by others
Introduction
Cryptosporidium parvum is a common zoonotic agent and has a wide host spectrum and geographical distribution, accounting for about 45% of human cryptosporidiosis cases worldwide1,2. There are extensive intraspecific variations in C. parvum populations. Currently, at least 14 C. parvum subtype families, including IIa, IIb, IIc, IId, IIe, IIf, IIg, IIh, IIi, IIk, IIl, IIm, IIn and IIo, have been identified on the basis of sequence analysis of the 60 KDa glykoprotein (GP60) gene1,2,3. Among them, IIa is the predominant subtype family in animals and humans worldwide whereas IId is another major zoonotic subtype family reported in Europe (Hungary, Germany, Portugal, Sweden, Ireland, Spain, Belgium, Romania, the United Kingdom, Netherlands, Slovenia and Serbia and Montenegro), Asia (Kuwait, Iran, Jordan, India, Malaysia and China), Egypt and Australia1,2,3,4,5,6,7. In contrast, IIc and IIe are anthroponotic subtype families. Other subtype families of C. parvum are occasionally seen in some animals or humans in the world2,4.
The life cycle of Cryptosporidium comprises a sexual phase during which gamete differentiation and fertilization occurs. This process can affect the population structure of this parasite because of the possibility of genetic recombination between genotypes. Experimental evidence has confirmed that recombinant genotypes can emerge from mixed infections8. In nature, panmictic population structure representing frequent genetic exchange was seen in human and bovine C. parvum isolates in Scotland, Ireland and the United States9,10,11. Meanwhile, epidemic structure was also noticed in C. parvum human isolates in Haiti and Scotland, in which genetic exchange and linkage equilibrium is masked by the expansion of genetically identical parasites12,13. In contrast, clonal population structure (indicates that multilocus subtypes are relatively stable in time and place) was observed in C. parvum human isolates in France13 and bovine C. parvum in Israel and Italy14,15. On the other hand, there is evidence that in one area, C. parvum can have an epidemic population structure in humans but a panmictic population structure in cattle12, or a clonal population structure in humans but an epidemic population structure in animals13. Therefore, the relative contribution of genetic recombination to the diversification of C. parvum in nature is complex and may depend on diverse factors including transmission intensity, host movement and host adaptation1.
In China, C. parvum has been detected in a small number of animals and humans. GP60 sequence analysis showed that all C. parvum isolates characterized thus far belonged to IId subtypes, including IIdA15G1 in rodents16, IIdA19G1 in cattle, humans and urban wastewater4,17,18,19. Previously, IIdA15G1 was detected in cattle, sheep and goats in Iran, Malaysia and Spain20,21,22,23 and in humans in Netherlands, Australia, Iran, Malaysia and India6,22,24,25,26. Likewise, IIdA19G1 was seen in cattle in Hungary and Sweden27,28 and in humans in Portugal and Sweden3,29. Thus, both IIdA15G1 and IIdA19G1 are considered zonootic subtypes.
Here we analyzed 111 C. parvum IId isolates from China, Sweden and Egypt by a multilocus sequence typing (MLST) technique using 12 microsatellite, minisatellite and single nucleotide polymorphism (SNP) loci. Our findings revealed that C. parvum IId subtypes had a clonal population structure and probably dispersed from Western Asia to other geographical regions.
Results
MLST subtypes and sequence polymorphism
All 111 specimens were successfully subtyped and 1–11 sequence types were found at the 12 gene loci. Among them, GP60 and DZ-HRGP had relatively higher sequence polymorphism, with 11 and 7 subtypes being identified, respectively. In contrast, all 111 specimens had the same sequence at the normally polymorphic RPGR locus. Altogether, these C. parvum IId specimens formed 25 MLST subtypes (Table S1).
Sequence data of all 12 loci were concatenated to make a multilocus gene of 7209 bp in length. The genetic diversity of sequences was analyzed using DnaSP 5.10.01. They had 135 polymorphic (segregating) sites and 12 haplotypes with a haplotype diversity Hd = 0.698 ± 0.039, nucleotide diversity Pi = 0.00692 and average number of nucleotide differences k = 47.296. The genetic diversity of Swedish isolates was higher than that of Chinese isolates (Table S2). Gene flow and genetic differentiation analysis showed significant differences among China, Egypt and Sweden populations (P < 0.001), with the pairwise fixation index (FST) of 0.811 to 0.919. Thus, the value of Nm representing gene flow was only 0.05 by both NST and FST estimates based on sequence data.
Higher genetic diversity level at the GP60 gene is observed in Western Asia
To estimate genetic diversity level of C. parvum IId subtypes, the GP60 locus representing the most polymorphic sequence identified thus far was used in this study. 43 GP60 sequences belonging to 15 subtypes were obtained from the GenBank database, including IIdA14G1, IIdA15G1, IIdA16G1, IIdA17G1, IIdA18G1, IIdA19G1, IIdA20G1, IIdA21G1, IIdA22G1, IIdA23G1, IIdA24G1, IIdA25G1, IIdA26G1, IIdA28G1 and IIdA29G1. DNA polymorphism was analyzed using DnaSP 5.10.01. The nucleotide diversity (Pi) of GP60 sequences was 0.00267 in Western Asia (Iran, Kuwait and Jordan), which was higher than that in other areas, including European countries, other Asian countries (China, India and Malaysia), Egypt and Australia (Table S3).
Phylogenetic analysis reveals a marked geographical separation of isolates
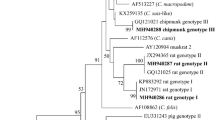
Neighbor-joining analysis of the concatenated sequences grouped the 25 MLST subtypes into three distinct lineages (Fig. 1). The lineages occupy strikingly distinct geographic distributions; lineage 3 occurred almost exclusively in China, with only Egyptian isolates (MLST-6) being included. In contrast, the Sweden isolates consisted of lineages 1 and 2. Likewise, the phylogenetic network analysis also produced the same result (Fig. S1).

Phylogenetic relationship among MLST subtypes of C. parvum IId isolates at 12 genetic loci as assessed by a neighbor-joining analysis of the concatenated nucleotide sequences, using distance calculated by the Kimura 2-parameter model.
The figure was generated using the softwares of Microsoft PowerPoint 2003 and Adobe Photoshop 8.0.1 after by the cycle model in MEGA version 4 opening the tree file.
LD analysis suggests clonal population structure
To examine LD within and between loci, we analyzed pairwise associations of polymorphism across the concatenated sequences. LD was estimated using the ZnS statistic, which calculates the average correlation among alleles in all pairs of polymorphic sites. ZnS was elevated not only for all isolates from the three countries but also for isolates within China or Sweden (Table 1). These high ZnS values indicated the C. parvum IId subtypes had a clonal population structure, suggesting the absence of recombination among lineages, which was supported by the D′ statistic (Fig. S2). In addition, the standardized index of association (ISA) analysis also confirmed this conclusion (Table S4).
Structure analysis produces three ancestral lineages
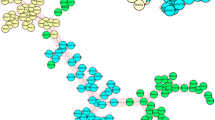
A Bayesian statistical approach was used to infer population substructure in allelic variation using the software STRUCTURE. The 111 C. parvum IId isolates derived from the admixture of three ancestral lineages (K = 3) (Fig. S3). Cryptosporidium parvum IId isolates from cattle, hamsters and horses in China and cattle in Egypt consisted of ancestral lineage III; whereas the 43 C. parvum IId isolates from cattle in Sweden formed ancestral lineages I and II (Fig. 2). Generally, Chinese isolates and Swedish isolates had different ancestral lineages. In contrast, the isolates from Egypt mainly showed the admixture of lineages I and III (Fig. 2).

Population structure inferred by Bayesian clustering using multilocus subtype information.
Ancestral population size of 3 (K = 3) was chosen as the best fit for the current data (Fig. S2). Upper labels are the ancestral populations; the geographic locations, MLST subtypes and hosts of the isolates are at the bottom. C = cattle, H = horse, M = mouse.
Discussion
Compared to length variations of polymorphic locus by multilocus typing (MLT) tools, multilocus sequence typing tools allow more definitive detection of sequence polymorphism and the inclusion of SNP makers1. In this study, 1–11 sequence types of C. parvum IId specimens were seen at each of the polymorphic loci, forming 25 MLST subtypes (Table S1). Some ZPT, ML2 and GP60 sequences were restricted to a single animal species, suggesting that they may be host adaptation30. However, whether these genetic differences represent different biologic characteristics is unclear.
ZnS statistic, D′ statistic and ISA values all confirmed that C. parvum IId subtypes had a clonal population structure, indicating that the MLST subtypes are relatively stable in time and place1. This population structure allows longitudinal tracking of the transmission of C. parvum IId subtypes in field studies. Previously, diverse C. parvum structures ranging from clonal, epidemic, to panmictic have been reported in a few studies12,13. However, most of these were conducted by MLG analyses of polymorphism of microsatellite and minisatellite loci in IIa and some IIc isolates. The actual genetic diversity of C. parvum isolates could be masked by the MLG tools, because many subtypes have the same length even though the sequences are different, such as HSP70, Mucin1, MSC6-7 and even GP60 genes1. Therefore, using high-resolution MLST tools in combination with more polymorphic loci are needed before firm conclusions can be reached on the population structure of different C. parvum subtype families.
Significant geographic segregation of MLST subtypes was observed in this study. The MLST subtypes from China, Egypt and Sweden differed from each other; even though they belonged to the same GP60 subtype IIdA19G1, MLST-1 and MLST-2 were seen in China whereas MLST-7 was noticed in Sweden (Table S1). Nevertheless, in Sweden, several MLST subtypes were detected in the same city or province and even within the same herd (Table S5). Thus, even in a clonal population structure, genetically distinct C. parvum IId subtypes can emerge within a group of hosts. The similar observation was also noticed in a previous study conducted in Israel and Turkey14. This may be partially attributed to the introduction of new animals into the herd or area via animal trade. Indeed, data showed that cattle trade was maintained at a considerable level in Sweden31. Therefore, frequent animal trade in Sweden would increase the heterogeneity of C. parvum IId subtypes on farms and the complexity of infections. Sequence analysis provided some evidence for the occurrence of mixed subtypes in Sweden, where a weak recombination event was seen by DnaSP 5.10.01 analysis (Rm = 2).
STRUCTURE analysis revealed three ancient lineages in the C. parvum IId specimens examined in this study (Fig. 2), which was supported by the phylogenetic relationship analyses (Fig. 1 and S1). However, the ancestry origin of the three ancient lineages was unclear. In addition, all 111 specimens had the same sequence at the RPGR gene which had 100% of identity with another zonootic C. parvum subtype IIaA15G1R1 (accession no. BX538350 and XM_627472), whereas shared only 93.6% of homology in comparison with the anthroponotic subtype IIcA5G3 (accession no. EU141711). Nevertheless, we do not know whether RPGR is an useful marker for distinguishing zonootic and anthroponotic subtypes and tracking the origin of C. parvum due to the absence of genetic data of other subtype families. Understanding the domestication history of domestic animals will help us to explore the origin of C. parvum. Modern cattle, sheep and goats are thought to have originated from Western Asia (especially in the Fertile Crescent) and introduced to the rest of the world during the last 10,500–15,000 years32,33,34,35,36. In addition, cattle, sheep and goats are the most common hosts for C. parvum1,2. Thus, C. parvum IId family probably dispersed from Western Asia to other areas as a result of cattle introduction, gradually forming various clonal populations because of host and geographical segregation. However, the divergence time of different subtype families of C. parvum is still unclear due to the absence of necessary references, such as molecular clock.
In conclusion, as expected, multiple MLST subtypes of C. parvum IId isolates were present in various animals examined in this study. Host-adaptation and significant geographical segregation of MLST subtypes were both observed among isolates from China, Sweden and Egypt. Cryptosporidium parvum IId isolates from China, Sweden and Egypt had a clonal population structure, shared the same RPGR sequence and consisted of three ancestral lineages. In addition, the GP60 sequences of C. parvum IId isolates from Western Asia had a higher nucleotide diversity in comparison with that in other areas. Therefore, these findings, in combination with the unique geographical distribution of C. parvum IId subtypes (Fig. 3 and 4) and the domestication history of cattle, sheep and goats, indicated that C. parvum IId subtypes were probably dispersed from Western Asia to other geographical regions. Nevertheless, more population genetic structure studies involving various C. parvum subtype families and high-resolution tools are needed to better elucidate the origin and spread of C. parvum in the world.

Ratio of the common C. parvum subtype families found in the world.
Numbers above bars represent numbers of samples subtyped at the GP60 locus. Data are based on studies published between 2004 and March 2013.

Distribution of C. parvum IId subtypes in the world.
The size of black circle indicates the number of isolates. Blue and red circles represent isolates from animals and humans, respectively. The figure was generated using the softwares of MapInfo Professional 7.0, Microsoft PowerPoint 2003 and Adobe Photoshop 8.0.1.
Methods
Cryptosporidium specimens
A total of 111 C. parvum IId specimens were used in this study including Chinese isolates from dairy cattle, horses and hamsters and Swedish and Egyptian isolates from dairy cattle (Table S1). Most of the specimens were identified in previous studies4,16,28, whereas the 10 bovine isolates from Egypt and six horse and five bovine isolates from China were diagnosed as C. parvum IId subtypes by DNA sequence analysis of the GP60 gene37.
Multilocus sequence typing
Twelve microsatellite, minisatellite and SNP loci were used in the present study. Primers and amplification conditions used in nested-PCR analysis of MSC6-7 (serine repeat antigen), RPGR (retinitis pigmentosa GTPase regulator), MSC6-5 (hypothetical trans-membrane protein), DZ-HRGP (hydroxyproline-rich glycoprotein), ZPT (zinc protease telomerase), CP56 (56 kDa trans-membrane protein), Chom3T (T-rich gene fragment), CP47 (47 kDa protein), HSP70 (70 kDa heat shock protein), Mucin1 (mucin-like protein), ML2 (possible sporozoite cystein-protein) and GP60 genes were previously described1. KOD-FX-Neo amplification enzyme (Toyobo Co. Ltd, Osaka, Japan) was used in PCR. 400 ng/μl of non-acetylated bovine serum albumin (Solarbio Co. Ltd, Beijing, China) was used in primary PCR to neutralize PCR inhibitors. The secondary PCR products were examined by agarose gel electrophoresis and visualized after GelRed™ (Biotium Inc., Hayward, CA) staining. The secondary PCR products were sequenced on an ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, USA), using the secondary primers and the Big Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems). The sequence accuracy was confirmed by bi-directional sequencing and by sequencing a new PCR product if necessary.
Phylogenetic relationship and DNA polymorphism analysis
Sequence alignment was made using the program ClustalX 1.83 (http://www.clustal.org/). Neighbor joining trees were constructed using the program Phylip 3.69, based on the evolutionary distances calculated by Kimura-2-parameter model. The reliability of these trees was assessed using the bootstrap analysis with 1,000 replicates. The phylogenetic network analysis was done using the median-joining algorithm as implemented in Network 4.611 based on the polymorphism of multilocus sequence data from 111 C. parvum IId isolates38. DNA polymorphism, pairwise FST values for the different populations and gene flow were calculated using DnaSP 5.10.0139.
Linkage disequilibrium
Sequences from 12 loci were combined in a single contig and analyzed for linkage disequilibrium (LD) across the entire composite sequence. LD analysis was conducted using software DnaSP 5.10.0139. D′ was calculated for all pairs of sites. Both the two-tailed Fisher's exact test and the χ2 test were used to determine significance of the associations between polymorphic sites. The average LD was estimated by using the ZnS statistic, which averages LD over all pairwise comparisons for S polymorphisms in N sequences. Alternatively, as a confirmation for the ZnS statistic, the standardized index of association (ISA) by Habould and Hudson (2000) was also used to assess the LD across all loci. The index and its probability under a null model of complete panmixia were calculated using LIAN version 3.540 with hypothesis testing by a parametric method.
Structure analysis
The subpopulations of C. parvum IId subtypes were determined based on the allelic data using STRUCTURE 2.3.3 by K-means partitional clustering and the admixture model41. Twenty simulation runs were conducted for each of K = 2 to 10 using a length of burn-in of 104 and 105 replicates of Markov chain Monte Carlo simulation. The average membership coefficients for the 20 simulation runs of a given K value were generated by CLUMPP v1.1.2 (windows version)42 and a graphical presentation of the average membership coefficients for each isolate was generated in Microsoft Excel. The most appropriate number of K was calculated using an ad hoc statistic-based approach implemented in software Structure Harvester v0.6.93, as described previously43.
Nucleotide sequence accession numbers
Representative nucleotide sequences were deposited in the GenBank under accession numbers KC823118 to KC823157.
References
Xiao, L. & Ryan, U. [Molecular epidemiology]. Cryptosporidium And Cryptosporidiosis [Fayer R., & Xiao L., (eds)] [119–150] (CRC Press and IWA Publishing, Boca Raton, 2008).
Xiao, L. Molecular epidemiology of cryptosporidiosis: an update. Exp. Parasitol. 124, 80–89 (2010).
Insulander, M. et al. Molecular epidemiology and clinical manifestations of human cryptosporidiosis in Sweden. Epidemiol. Infect. 141, 1009–1020 (2013).
Wang, R. J. et al. Characteristics of Cryptosporidium transmission in preweaned dairy cattle in henan, China. J. Clin. Microbiol. 49, 1077–1082 (2011).
Amer, S. et al. Identity and public health potential of Cryptosporidium spp. in water buffalo calves in Egypt. Vet. Parasitol. 191, 123–127 (2012).
Iqbal, A., Lim, Y. A., Surin, J. & Sim, B. L. High diversity of Cryptosporidium subgenotypes identified in Malaysian HIV/AIDS individuals targeting gp60 gene. PLoS One 7, e31139 (2012).
Imre, K. et al. Zoonotic Cryptosporidium parvum in Romanian newborn lambs (Ovis aries). Vet. Parasitol. 191, 119–122 (2013).
Feng, X., Rich, S. M., Tzipori, S. & Widmer, G. Experimental evidence for genetic recombination in the opportunistic pathogen Cryptosporidium parvum. Mol. Biochem. Parasitol. 119, 55–62 (2002).
Mallon, M. et al. Population structures and the role of genetic exchange in the zoonotic pathogen Cryptosporidium parvum. J. Mol. Evol. 56, 407–417 (2003).
Herges, G. R. et al. Evidence that Cryptosporidium parvum populations are panmictic and unstructured in the Upper Midwest of the United States. Appl. Environ. Microbiol. 78, 8096–8101 (2012).
De Waele, V. et al. Panmictic structure of the Cryptosporidium parvum population in irish calves: influence of prevalence and host movement. Appl. Environ. Microbiol. 79, 2534–2541 (2013).
Mallon, M. E., Macleod, A., Wastling, J. M., Smith, H. & Tait, A. Multilocus genotyping of Cryptosporidium parvum Type 2, population genetics and sub-structuring. Infect. Genet. Evol. 3, 207–218 (2003).
Ngouanesavanh, T. et al. Cryptosporidium population genetics: evidence of clonality in isolates from France and Haiti. J. Eukaryot. Microbiol. 53(Suppl), 33–36 (2006).
Tanriverdi, S. & Widmer, G. Differential evolution of repetitive sequences in Cryptosporidium parvum and Cryptosporidium hominis. Infect. Genet. Evol. 6, 113–122 (2006).
Drumo, R. et al. Evidence of host-associated populations of Cryptosporidium parvum in Italy. Appl. Environ. Microbiol. 78, 3523–3529 (2012).
Lv, C. C. et al. Cryptosporidium spp. in wild, laboratory and pet rodents in china: prevalence and molecular characterization. Appl. Environ. Microbiol. 75, 7692–7699 (2009).
Li, N. et al. Molecular surveillance of Cryptosporidium spp., Giardia duodenalis and Enterocytozoon bieneusi by genotyping and subtyping parasites in wastewater. PLoS Negl. Trop. Dis. 6, e1809 (2012).
Zhang, W. Z. et al. Distribution and genetic characterizations of Cryptosporidium spp. in pre-weaned dairy calves in Northeastern China's Heilongjiang Province. PLoS One 8, e54857 (2013).
Wang, L. et al. Zoonotic Cryptosporidium species and Enterocytozoon bieneusi genotypes in HIV-positive patients on antiretroviral therapy. J. Clin. Microbiol. 51, 557–563 (2013).
Quilez, J. et al. Cryptosporidium species and subtype analysis from dairy calves in Spain. Parasitology 135, 1613–1620 (2008).
Quilez, J. et al. Cryptosporidium genotypes and subtypes in lambs and goat kids in Spain. Appl. Environ. Microbiol. 74, 6026–6031 (2008).
Nazemalhosseini-Mojarad, E. et al. Subtype analysis of Cryptosporidium parvum and Cryptosporidium hominis isolates from humans and cattle in Iran. Vet. Parasitol. 179, 250–252 (2011).
Muhid, A., Robertson, I., Ng, J. & Ryan, U. Prevalence of and management factors contributing to Cryptosporidium sp. infection in pre-weaned and post-weaned calves in Johor, Malaysia. Exp. Parasitol. 127, 534–538 (2011).
Wielinga, P. R. et al. Molecular epidemiology of Cryptosporidium in humans and cattle in The Netherlands. Int. J. Parasitol. 38, 809–817 (2008).
Ng, J., MacKenzie, B. & Ryan, U. Longitudinal multi-locus molecular characterisation of sporadic Australian human clinical cases of cryptosporidiosis from 2005 to 2008. Exp. Parasitol. 125, 348–356 (2010).
Ajjampur, S. S. et al. Multisite study of cryptosporidiosis in children with diarrhea in India. J. Clin. Microbiol. 48, 2075–2081 (2010).
Plutzer, J. & Karanis, P. Genotype and subtype analyses of Cryptosporidium isolates from cattle in Hungary. Vet. Parasitol. 146, 357–362 (2007).
Silverlas, C., Bosaeus-Reineck, H., Naslund, K. & Bjorkman, C. Is there a need for improved Cryptosporidium diagnostics in Swedish calves? Int. J. Parasitol. 43, 155–161 (2013).
Alves, M., Xiao, L., Antunes, F. & Matos, O. Distribution of Cryptosporidium subtypes in humans and domestic and wild ruminants in Portugal. Parasitol. Res. 99, 287–292 (2006).
Xiao, L. et al. Host adaptation and host-parasite co-evolution in Cryptosporidium: implications for taxonomy and public health. Int. J. Parasitol. 32, 1773–1785 (2002).
Widgren, S. & Frossling, J. Spatio-temporal evaluation of cattle trade in Sweden: description of a grid network visualization technique. Geospat. Health 5, 119–130 (2010).
Gotherstrom, A. et al. Cattle domestication in the Near East was followed by hybridization with aurochs bulls in Europe. Proc. Biol. Sci. 272, 2345–2350 (2005).
Beja-Pereira, A. et al. The origin of European cattle: evidence from modern and ancient DNA. Proc. Natl. Acad. Sci. U. S. A. 103, 8113–8118 (2006).
Fernandez, H. et al. Divergent mtDNA lineages of goats in an Early Neolithic site, far from the initial domestication areas. Proc. Natl. Acad. Sci. U. S. A. 103, 15375–15379 (2006).
Chessa, B. et al. Revealing the history of sheep domestication using retrovirus integrations. Science 324, 532–536 (2009).
Ajmone-Marsan, P., Garcia, J. F. & Lenstra, J. A. On the origin of cattle: How aurochs became cattle and colonized the world. Evolutionary Anthropology: Issues, News and Reviews 19, 148–157 (2010).
Sulaiman, I. M. et al. Unique endemicity of cryptosporidiosis in children in Kuwait. J. Clin. Microbiol. 43, 2805–2809 (2005).
Bandelt, H. J., Forster, P. & Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Rozas, J., Sanchez-DelBarrio, J. C., Messeguer, X. & Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497 (2003).
Haubol, B. & Hudson, R. R. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–848 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet. Resour. 4, 359–361 (2012).
Acknowledgements
This study was supported in part by the State Key Program of National Natural Science Foundation of China (31330079), the National Natural Science Foundation of China (U1204328, 31172311, 31302079), the International Cooperation and Exchange Funds of the National Natural Science Foundation of China (31110103901) and the Key National Science and Technology Specific Projects (2012ZX10004-220).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: L.X.Z., L.H.X. and R.J.W. Performed the experiments: R.J.W., F.C.J., M.Q. and H.J.D. Analyzed the data: R.J.W., G.Q.L., C.C.L., Z.F.Z., H.L.W. and Y.R.S. Contributed reagents/materials/analysis tools: C.A., C.B., S.A., A.Q.L., Y.Y.F. and C.S.N. Wrote the paper: R.J.W., L.X.Z. and L.H.X.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wang, R., Zhang, L., Axén, C. et al. Cryptosporidium parvum IId family: clonal population and dispersal from Western Asia to other geographical regions. Sci Rep 4, 4208 (2014). https://doi.org/10.1038/srep04208
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04208
This article is cited by
-
Prevalence and molecular characterization of Cryptosporidium spp. in dairy and beef cattle in Shanxi, China
Parasitology Research (2024)
-
CRISPR/Cas12a-based on-site diagnostics of Cryptosporidium parvum IId-subtype-family from human and cattle fecal samples
Parasites & Vectors (2021)
-
Circular RNA ciRS-7 affects the propagation of Cryptosporidium parvum in HCT-8 cells by sponging miR-1270 to activate the NF-κB signaling pathway
Parasites & Vectors (2021)
-
Population structure and geographical segregation of Cryptosporidium parvum IId subtypes in cattle in China
Parasites & Vectors (2020)
-
Prevalence and molecular characterization of Cryptosporidium spp. and Giardia duodenalis in deer in Henan and Jilin, China
Parasites & Vectors (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
