


















« Prev Next »

Chromatin
If the DNA strand in a single human cell were stretched out, it would measure several meters in length. Thus, this strand must coil and kink many times over in order to fit into the cell's nucleus, which has a diameter only about one-tenth that of a human hair. To achieve this highly condensed form, the DNA winds itself around proteins called histones, thereby forming a complex known as chromatin.
Interestingly, chromatin not only serves as a way to condense DNA within the cellular nucleus, but also as a way to control how that DNA is used. In particular, within eukaryotes, specific genes are not expressed unless they can be accessed by RNA polymerase and proteins known as transcription factors. In its default state, the tight coiling that characterizes chromatin structure limits the access of these substances to eukaryotic DNA. Therefore, a cell's chromatin must "open" in order for gene expression to take place. This process of "opening" is called chromatin remodeling, and it is of vital importance to the proper functioning of all eukaryotic cells. In recent years, researchers have discovered a great deal about chromatin remodeling, including the roles that different protein complexes, histone variants, and biochemical modifications play in this process. However, a great deal remains to be learned before chromatin remodeling is fully understood.
Chromatin Remodeling at a Glance
Various molecules called chromatin remodelers provide the mechanism for modifying chromatin and allowing transcription signals to reach their destinations on the DNA strand. Understanding the nature and processes of these cellular construction workers remains an active area of discovery in genetic research.
Currently, investigators know that chromatin remodelers are large, multiprotein complexes that use the energy of ATP hydrolysis to mobilize and restructure nucleosomes. Recall that nucleosomes wrap 146 base pairs of DNA in approximately 1.7 turns around a histone-octamer disk, and the DNA inside each nucleosome is generally inaccessible to DNA-binding factors. Remodelers are thus necessary to provide access to the underlying DNA to enable transcription, chromatin assembly, DNA repair, and other processes. Just how remodelers convert the energy of ATP hydrolysis into mechanical force to mobilize the nucleosome, and how different remodeler complexes select which nucleosomes to move and restructure, remains unknown, however.
Remodelers are partitioned into five families, each with specialized biological roles. Nonetheless, all remodelers contain a subunit with a conserved ATPase domain. In addition to the conserved ATPase, each remodeler complex also possesses unique proteins that specialize it for its unique biological role. However, because all remodelers move nucleosomes and all such movement is ATP dependent, mobilization is most likely a property of the conserved ATPase subunit.
The ATPase domains of remodelers are similar in sequence and structure to known DNA-translocating proteins in viruses and bacteria. Recent evidence from the SWI/SNF and ISWI remodeler families has also revealed that remodeler ATPases are directional DNA translocases that are capable of the directional pumping of DNA. But how is this property applied to nucleosomes? It seems that the ATPase binds approximately 40 base pairs inside the nucleosome, from which location it pumps DNA around the histone-octamer surface. This enables the movement of the nucleosome along the DNA, thus permitting the exposure of the DNA to regulatory factors.
The
additional domains and proteins that are attached to the ATPase are important
for nucleosome selection, and they also help regulate ATPase activity. These
attendant proteins bind to histones and nucleosomal DNA, and their binding to
these molecules is affected by the histone modification state. The modification
state helps determine whether the nucleosome is an appropriate substrate for a
remodeler complex (Saha et al.,
2006), as discussed later in this article.
Nucleosomes, Histones, and Gene Transcription
Chromatin structure is highly complex and impressively
dynamic. Scientists now realize that the nucleosome, which was once thought to be static, actually plays an integral role in directing some elements of transcriptional specification (Figure 1).
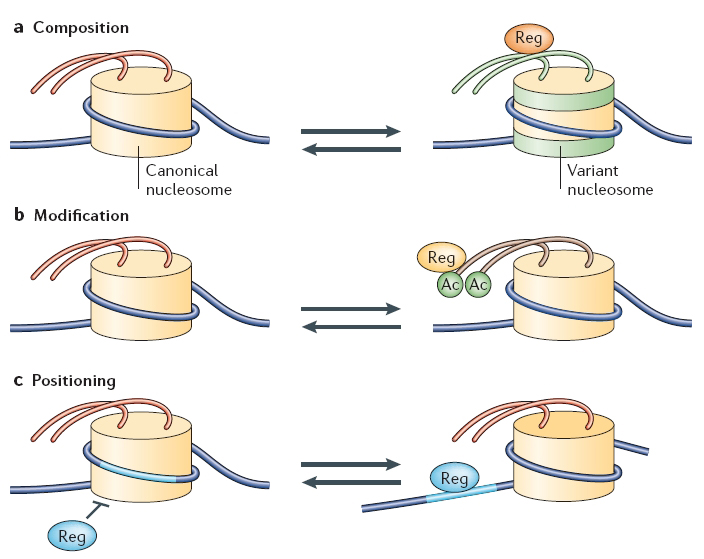
Figure 1: Dynamic properties of nucleosomes
(A) Remodelling complexes of the SWR1 family can remove the canonical H2A-H2B dimers and replace them with Htz1-H2B dimers (indicated in green), forming a variant nucleosome with unique tails that might bind unique regulatory proteins (Reg). (B) Nucleosome modification (only acetylation [Ac] is depicted for simplicity) allows the binding of regulatory factors, which have specialized domains (bromodomains) that recognize acetylated histone tails. (C) Nucleosome repositioning allows the binding of a regulatory factor to its site on nucleosomal DNA (light-blue segment). Remodellers are necessary to provide rapid access to nucleosomal DNA by sliding of the octamer along the DNA. Alternatively, the site might be accessed on the surface through the generation of a DNA wave or through histone-octamer ejection (not shown).
© 2006 Nature Publishing Group Saha, A. et al. Chromatin remodelling: the industrial revolution of DNA around histones. Nature Reveiws Molecular Cell Biology 7, 437–447 (2006) doi:10.1038/nrm1945. All rights reserved.
A nucleosome core particle consists of eight histone proteins (two each of H2A, H2B, H3 and H4) and 146 base pairs of double-stranded DNA. The composition of nucleosomes is not set in stone, however. Indeed, canonical histones can themselves be replaced by histone variants or modified by specific enzymes, thereby making the surrounding DNA more or less accessible to the transcriptional machinery.
So far, a number of histone variants have been found and localized to specific areas of chromatin. For instance, H2A.Z is a variant of H2A and is often enriched near relatively inactive gene promoters. Interestingly, H2A.Z does not take its place during replication when the chromatin structure is established. Instead, the chromatin remodeling complex SWR1 catalyzes an ATP-dependent exchange of H2A in the nucleosome for H2A.Z (Wu et al., 2005).
CENP-A is another known histone variant that has been found to be associated with centromeres. Originally localized to the centromere through immunofluorescence studies, CENP-A was believed to be involved in centromeric activity during cell division. But, once the CENP-A protein was isolated and sequenced, it was shown to have sequence homology to H3, suggesting that CENP-A actually replaces canonical H3 near the centromere. Some experiments suggest that these variant histones that occur in particular areas of the genome may assist in the specific regulation of chromatin behavior and gene transcription from these areas (Li et al., 2005).
Histone Modification and the Histone Code
Histone sequences are highly conserved. The diagram in Figure 2 shows a typical chromatin fiber, with the blue cylinders representing histones. Extending from each of the histones is a "tail," called the N-terminal tail because proteins have two ends--an N terminus and C terminus. Here, the C terminus forms a globular domain that is packaged into the nucleosome. The other end of the histone is more flexible and capable of interacting more directly with DNA and the different proteins within the nucleus.

Figure 2: The N terminus.
(A) General chromatin organization. Like other histone "tails," the N terminus of H3 (red) represents a highly conserved domain that is likely to be exposed or extend outwards from the chromatin fiber. A number of distinct post-translational modifications are known to occur at the N terminus f H3 including acetylation (green flag), phosphorylation (grey circle), and methylation (yellow hexagon). Other modifications are known and may also occur in the globular domain. (B) The N terminus of human H3 is shown in single-letter amino-acid code. For comparison, the N termini of human CENP-A, a centromere-specific H3 variant, and human H4, the nucleosomal partner to H3, are shown. Note the regular spacing of acetylatable lysines (red), and potential phosphorylation (blue) and methylation (purple) sites. The asterisk indicates the lysine residue in H3 that is known to be targeted for acetylation as well as for methylation; lysine 9 in CENP-A (bold) may also be chemically modified (see text). The above depictions of chromatin structure and H3 are schematic; no attempt has been made to accurately portray these structures.
© 2014 Nature Publishing Group Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 42 (2000). All rights reserved. 
Specifically, histone modification involves covalent bonding of various functional groups to the free nitrogens in the R-groups of lysines in the N-terminal tail. Early research has linked differing levels of acetylation and methylation on the histones to altered rates of DNA transcription (Turner, 2005). While the most common additions are acetylation and methylation of lysine residues, many more types of modifications have also been observed, including phosphorylation, a common posttranslational modification. The different types of modifications, which have been called the "histone code," are put in place by a variety of different enzymes, many of which have yet to be fully characterized. Thus, the story of the remodeling machinery continues to be told through a variety of experiments, and much remains to be revealed.
Nucleosome Positioning and Reorganization
Because eukaryotic DNA is tightly wrapped around nucleosomes and the positive charges of the histones tightly bind the negative charges of the DNA, nucleosomes essentially act as a physical barrier to transcription factors that need to bind to certain regions of DNA. However, specific acetylations can remove the positive charge on the lysine amino group that is acetylated, so the nucleosome "loses its grip" on the DNA. This modification results in a loosening of the coil.
Other remodeling enzyme complexes actually slide the nucleosomes along the DNA to clear them from the promoter regions (Cosgrove et al., 2004). In this case, the remodeling enzymes use the energy from ATP to regulate nucleosome movement. For example, prior to transcription in yeast, one of the major types of chromatin remodeling machines, called the SWI/SNF and SAGA histone acetylase complex, is recruited to the yeast HO gene promoter by the SWI5 activator. Activator-dependent chromatin modification then moves the nucleosome out of the way so that RNA polymerase II can reach the promoter regions of the DNA (Struhl, 1999).
Chromatin remodeling activity by SWI/SNF or other remodeling machines can also be required for recruiting additional chromatin remodeling activity to the site, as well as additional downstream sites. Modifications at a promoter can occur in multiple steps that are independently regulated, and additional modifications can occur stepwise stretching from the point of the first modification along the DNA strand in a downstream direction toward the promoter. These modifications open up an elongated region of active chromatin and allow for a wide range of intermediate, transcriptionally inactive states for the eukaryotic promoter. Promoters can also be poised with RNA polymerase bound but not elongating the mRNA; in yeast, up to 15% of sites have such stalled transcription. Changes in gene expression during the specific developmental stages of an organism or cell coincide with fluctuations in the levels of each of the specific protein complexes involved in chromatin remodeling (Struhl, 1999).
Signaling Function of Remodeled Chromatin
Histone modification can open chromatin, thus permitting selective binding of transcription factors that, in turn, recruit RNA polymerase II (Turner, 2005). Varying levels and types of histone modifications have been shown to correlate with levels of chromatin activation. For example, one group of researchers used antibody-based immunoprecipitation studies to determine that acetylation of histone H3 and methylation at lysine residue K4 appeared to coincide with each other. They also coincided during transcriptional activation in chicken embryos, while methylation at lysine residue K9 marked inactive chromatin.
DNA Methylation
Another means by which transcription is controlled is through methylation of the DNA strand itself. Not to be confused with histone methylation, methylation of the DNA strand involves cytosine bases of eukaryotic DNA being converted to 5-methylcytosine, resulting in the repression of transcription, particularly in vertebrates and plants. The altered cytosine residues are usually immediately adjacent to a guanine nucleotide, resulting in two methylated cytosine residues set diagonally to one another on opposing DNA strands (Figure 3).

Figure 3: DNA methylation and gene silencing
In early embryogenesis, DNA is largely devoid of methylation (top left). Post implantation, de novo methylation begins (red circles), mediated primarily by DNA (cytosine-5-)-methyltransferase-3alpha (DNMT3A) and -3beta (DNMT3B) (top). When methylation affects CpG islands, methyl-binding proteins trigger a silencing cascade (activity illustrated by green stars) whereby histone H3K9 is sequentially deaceylated and then methylated, allowing heterochromatin protein 1 (HP1) to bind; eventually resulting in closed chromatin (bottom right). After DNA replication, newly synthesized DNA (in green) is unmethylated. However, DNMT1 rapidly scans DNA and deposits methyl groups on newly synthesized DNA, opposite methyl groups present on the old DNA strand. This results in faithful replication of methylation patterns (bottom left) and the maintenance of silencing. Adult patterns of methylation are erased by epigenetic reprogramming in early embryogenesis (top left).
© 2004 Nature Publishing Group Issa, J. CpG island methylator phenotype in cancer. Nature Review Cancer 4, 989 (2004). All rights reserved.
Heavily methylated regions of DNA with elevated concentrations of these so-called CpG groups are often found near transcription start sites. In an interestingly coordinated process, proteins that bind to methylated DNA also form complexes with proteins involved in deacetylation of histones. Therefore, when the DNA is in a methylated state, nearby histones are deacetylated, resulting in compact, semipermanently silent chromatin. Likewise, demethylated DNA does not draw deacetylating enzymes to the histones, but it often attracts histone acetyltransferases, allowing histones to remain acetylated and promoting transcription.
Summary
Storage of eukaryotic DNA in small, compact nuclei requires that this DNA be tightly coiled and compacted in the form of chromatin. However, the structure of chromatin also appears to serve a second, possibly more important role, in that it gives eukaryotic cells the capability to exert complex levels of control over gene expression.
As described throughout this article, chromatin and the DNA sequences it contains are constantly undergoing modifications, thereby periodically exposing different regions of DNA to transcription factors and RNA polymerases. The cumulative effects of these changes are various states of transcriptional control and the ability of eukaryotic cells to turn genes on and off as needed. This complexity provides eukaryotes with a means of making the most of a relatively small number of genes. However, much research remains to be performed before investigators precisely understand how the many mechanisms of chromatin remodeling operate, as well as how they work together to result in the complex patterns of gene expression characteristic of eukaryotic cells.
References and Recommended Reading
Cosgrove, M., et al. Regulated nucleosome mobility and the histone code. Nature Structural and Molecular Biology 11, 1037–1043 (2004) (link to article)
Issa, J. P. CpG island methylator phenotype in cancer. Nature Reviews Cancer 4, 988–993 (2004) (link to article)
Jenuwein T, Allis CD. Translating the histone code. Science 293, 1074-1080 (2001)
Lall, S. Primers on chromatin. Nature Structural and Molecular Biology 14, 1110–1115 (2007) (link to article)
Li, B., et al. Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proceedings of the National Academy of Sciences 102, 18385–18390 (2005)
Pierce, B. Genetics: A Conceptual Approach, 2nd ed. (New York, Freeman, 2004)
Sadava, D., et al. Life: The Science of Biology, 8th ed. (New York, Freeman, 2006)
Saha, A., et al. Chromatin remodelling: The industrial revolution of DNA around histones. Nature Reviews Molecular Cell Biology 7, 437–447 (2006) (link to article)
Strahl, B., & Allis, D. C. The language of covalent histone modifications. Nature 403, 41–45 (2000) doi:10.1038/47412 (link to article)
Struhl, K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell 98, 1–4 (1999)
Turner, B. Reading signals on the nucleosome with a new nomenclature for modified histones. Nature Structural and Molecular Biology 12, 110–112 (2005) (link to article)
Wu, W. H., et. al. SWC2 is a widely conserved H2AZ-binding module essential for ATP-dependent histone exchange. Nature Structural and Molecular Biology 12, 1064–1071 (2005) doi:10.1038/nsmb1023 (link to article)
Zhang, Y., & Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes and Development 15, 2343-60 (2001)










