
Abstract
Background:
The genetic basis of dysfunctional immune responses in necrotizing enterocolitis (NEC) remains unknown. We hypothesized that variants in nucleotide binding and oligomerization domain (NOD)-like receptors (NLRs) and autophagy (ATG) genes modulate vulnerability to NEC.
Methods:
We genotyped a multi-center cohort of premature infants with and without NEC for NOD1, NOD2, ATG16L1, CARD8, and NLRP3 variants. Chi-square tests and logistic regression were used for statistical analysis.
Results:
In our primary cohort (n = 1,015), 86 (8.5%) infants developed NEC. The A allele of the ATG16L1 (Thr300Ala) variant was associated with increased NEC (AA vs. AG vs. GG; 11.3 vs. 8.4 vs. 4.8%, P = 0.009). In regression models for NEC that adjusted for epidemiological confounders, GA (P = 0.033) and the AA genotype (P = 0.038) of ATG16L1 variant were associated with NEC. The association between the A allele of the ATG16L1 variant and NEC remained significant among Caucasian infants (P = 0.02). In a replication cohort (n = 259), NEC rates were highest among infants with the AA genotype but did not reach statistical significance.
Conclusion:
We report a novel association between a hypomorphic variant in an autophagy gene (ATG16L1) and NEC in premature infants. Our data suggest that decreased autophagy arising from genetic variants may confer protection against NEC.
Similar content being viewed by others


Main
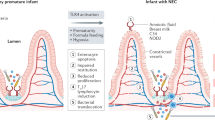
Necrotizing enterocolitis (NEC) is a form of intestinal inflammatory disease that develops in 5–12% of premature infants with a mortality of 20–35% (1,2). Infants with NEC present initially with abdominal distension, feeding intolerance, and bloody stools but often progress to systemic disease with multi-organ involvement and shock (2,3). It is generally accepted that risk factors, which portend intestinal mucosal injury or aberrant intestinal such as prematurity, hyperosmolar feeds, ischemia, formula milk feeding, and antibiotics are associated with NEC (1,2,3). However, the presence of these risk factors is neither necessary nor sufficient for NEC occurrence or severity in preterm infants. Recent studies have therefore attempted to determine genetic factors that contribute to the variation in liability to NEC observed in the presence of known risk factors (4,5). A major focus of investigation has been the immune system, as animal data and human studies indicate that aberrant activation of innate immune responses contributes to inflammation and mucosal injury in NEC (1,6,7). In this multi-center study, we investigated the relationship between sequence variants in the nucleotide oligomerization domain (NOD)-like receptor (NLRs) genes and NEC in premature infants.
NLRs are a family of evolutionary conserved intracellular immune receptors that recognize pathogen-associated molecular patterns and danger-associated molecular patterns in immune and nonimmune cells (8,9,10). They play important roles in regulating immunity, apoptosis, and autophagy, and their function/dysfunction has been extensively studied in the context of gastrointestinal tract homeostasis and inflammatory bowel disease (8,9,10). NOD1 and NOD2 recognize bacterial moieties produced during the synthesis or degradation of the bacterial cell wall component, peptidoglycan. NOD1 senses D-glutamyl-meso-diaminopimelic acid primarily from Gram-negative bacteria, while NOD2 senses muramyl dipeptide, present ubiquitously in Gram-positive and Gram-negative bacteria. NOD1- and NOD2-dependent bacterial recognition activates proinflammatory signaling through the NF-κB and MAPK pathways (9,10). Additionally, NOD1 and NOD2 signaling primes bacterial autophagy, a process by which bacteria are marked for autophagolysosomal clearance by recruiting the key autophagy protein Autophagy related 16-Like 1 (ATG16L1) (10,11). In contrast with NOD1 and NOD2, other members of the NLR family, such as NLRP3, regulate inflammasome function (11,12). The NLRP3 inflammasome is a multimeric protein complex that assembles in the cytoplasm upon sensing pathogen-associated molecular patterns and nonmicrobial stimuli such as uric acid, ATP, and reactive oxygen species (11,13). Activation of the NLRP3 complex initiates pro-Caspase-1 (apoptotic factor) and pro-IL-1β processing with subsequent release of active proteins. The Caspase Recruitment Domain containing protein-8 (CARD8) interacts with NLRP3 and NOD2 to inhibit inflammation (12,14).
Genetic variants in NLRs modulate susceptibility to inflammatory bowel disease (IBD), Crohn’s disease, and immunodeficiency in humans (12,13,15,16,17,18). Specifically, loss or gain of function variants in NOD1 (rs6958571), NOD2 (rs2066844), ATG16L1 (rs2241880), CARD8 (rs2043211), and NLRP3 (rs4353135, rs6672995, and rs35829419) have been associated with altering susceptibility or severity of IBD in adults and children (13,14,15,19,20). Given the general acceptance of: (i) dysregulated innate immune responses in NEC, (ii) the wide array of bacterial pathogen-associated molecular patterns and danger-associated molecular patterns in the premature infant gut that can be sensed by NLRs, and (iii) the known association of NLR genetic variants with IBD, a disease characterized by dysregulated intestinal immune responses, we hypothesized that functional genetic variants in NLR pathway genes will be associated with NEC in premature infants. In this study, we tested this hypothesis in a large cohort of premature infants.
Methods
Recruitment of Study Subjects
Premature infants were recruited from neonatal intensive care units at Children’s Hospital of Wisconsin (Milwaukee, WI), St. Joseph’s Hospital (Milwaukee, WI), Kosair’s Children’s Hospital (Louisville, KY), Children’s Hospitals and Clinics of Minnesota (Minneapolis, MN), and the University of Alabama at Birmingham (Birmingham, AL) after institutional review board approval at respective centers. After informed consent, 0.5 ml of blood was collected in coded sample containers, and shipped to Children’s Hospital of Wisconsin where DNA extraction and genotyping was done. Clinical and epidemiological data from infants recruited at these centers were deidentified and assigned a study code linked to their deidentified blood samples. Additional premature infant DNA samples were obtained from subjects recruited through institutional review board-approved studies at University of Iowa Children’s Hospital (Iowa City, IA) and BC Children’s & Women’s Health Centre (Vancouver, Canada). Sample recruited through a collaborative effort at Yale-New Haven Children’s Hospital (New Haven, CT), as part of the Neonatal Genetics Group (listed in the acknowledgment section) was used for a replication cohort. The study was approved by the Human Investigation Committee at Yale University as well as the respective Institutional Review Boards at the collaborating institutions. For infants from these sites, deidentified DNA samples and clinical data were used for studies and analysis.
Eligibility Criteria
Infants born with a gestational age ≤ 35 wk admitted to the participating centers were eligible. Infants with major congenital anomalies of the heart, gastro-intestinal tract, renal or respiratory tract were excluded.
Case definition. NEC was defined as per modified Bell’s criteria (21). Infants with stage I NEC were analyzed as cases. Infants who had surgery for NEC were also identified. Spontaneous ileal perforation was distinguished from NEC based on age of diagnosis, feeding history, and surgical data. These infants were excluded as NEC cases.
Selection of SNPs
Candidate NLR Single Nucleotide polymorphism (SNPs) were identified based on whether: (i) associations with inflammatory bowel disease or Crohn’s disease have been reported, (ii) variants had a functional effect, and (iii) variants had a minor allele frequency > 2% in the Caucasian population.
Laboratory Procedure
Genomic DNA was extracted from blood samples using the FlexiGene DNA kit (Qiagen, Valencia, CA) and stored at 4 °C. To genotype the NOD1 (rs6958571), NOD2 (rs2066844), ATG16L1 (rs2241880), CARD8 (rs2043211), and NLRP3 (rs4353135, rs6672995, and rs35829419) variants, we performed a 5′ nuclease Taqman assay (Applied Biosystems, Foster City, CA) using custom or predesigned TaqMan SNP Genotyping Assay probes (ABI, Foster City, CA) as per the manufacturer’s instructions (22). The principle of the assay involves amplification of the genomic region of interest followed with ligation with allele-specific probes that emit a distinct fluorescent signal specific to the variant or reference allele. Samples were analyzed on ABI HT7900 with SDS 2.3 software package (probes available on request). Genotyping was done by personnel blinded to clinical outcomes. Quality control. 10% of the samples were regenotyped by an independent technician blinded to prior results. There was >99% concordance for all samples.
Statistical Analysis
Chi-square analyses were used for comparisons between dichotomous clinical variables and NEC outcomes. Gestational age (GA) and birth weight were compared between groups using the Wilcoxon-Mann-Whitney rank sum test. Associations between genetic variants and GA were examined using the Kruskal-Wallis test. We used an additive genetic risk model to analyze relationships between SNPs and NEC outcomes. Variant allele frequencies were compared among infants with and without NEC using the Pearson’s Chi-square test or Fisher’s exact test. The Cochran-Armitage trend test was used to examine the additive risk associated with risk alleles. In prespecified a priori analysis, relationships between SNPs and NEC would be examined in Caucasian (CAU) and African American (AA) infants independently. We also examined the association between NLR variants and surgical NEC in our cohort.
Power. Assuming a NEC incidence of 8% in our primary cohort, we estimated that a sample size of ~ 950 infants would give us 80% power with a P = 0.05 to detect a 10–20 % difference in the prevalence of the variant allele between infants with and without NEC. To control for potential epidemiological confounders, we analyzed data using logistic regression with backward elimination where the probability of removal was set at P ≥ 0.05. In this model, birth variables (GA, birth weight, clinical chorioamnionitis, antenatal steroid exposure, race, sex) along with NLR SNPs were examined for association with NEC. Variables were removed from the model in a stepwise fashion till only those associated with NEC (P < 0.05) remained. Risk factors for severe NEC were modeled in a similar fashion. The Hosmer and Lemeshow Goodness-of-Fit test was used to assess the degree of fit in regression models. SPSS 21.0 (SPSS, Chicago, IL) and SAS 9.2 (SAS, NC) were used for data analysis.
Results
Epidemiology of NEC in Our Primary Cohort
We recruited 1,015 (n = 1015) premature infants with a gestational age (GA) ≤ 35 wk from multiple centers for our primary cohort. Eighty six infants (8.5%) were diagnosed with NEC in our primary cohort. Birth weight, antenatal steroid use, proportion of males, type of feeding, rates of medically treated PDA, and clinical chorioamnionitis were not significantly different among infants with or without NEC ( Table 1 ). NEC rates decreased with advancing GA (P = 0.006). Data on clinical chorioamnionitis and type of feeding were not available for 385 infants. We noticed a nonsignificant trend toward increased NEC among infants of African American descent (23/197 (11.7%) vs. 63/818 (7.7%), P = 0.07), and among infants with a birth weight <1,000 g (P = 0.07).
NLR Variants and NEC in the Primary Cohort
Hardy-Weinberg equilibrium was confirmed at the seven loci investigated. Genotype frequencies of NLR variants among infants without NEC, all NEC, and surgical NEC are shown in Table 2 . Genotyping data were not obtained in nine subjects for one or more variants because of inadequate sample. Using an additive risk model, we found no association between the NOD1 (rs6958571), NLRP3 (rs4353135, rs6672995, rs35829419), NOD2 (rs2066844), and CARD8 (rs2043211) SNPs and NEC in our cohort ( Table 2 ). The ATG16L1 (rs2241880, Thr300Ala) variant was associated with NEC (P = 0.009, Cochran-Armitage trend test) in our cohort. NEC (All NEC) prevalence was highest among infants with the AA genotype and decreased proportionately with addition of the G allele ( Figure 1a ). The AA, AG, and GG genotype frequencies of the ATG16L1 SNP did not vary with gestational age at birth in the overall cohort (median; 27 vs. 28 vs. 27 wk, P = 0.70), or in infants with NEC. Compared to infants with the GG genotype, infants with the AA genotype had a 2.5-fold increase in NEC (OR = 2.5, 95% CI = 1.2–5.6, P = 0.01). In logistic regression models for NEC that included clinical variables and SNPs, only gestational age (OR = 0.9; 95% CI = 0.83–0.99, P = 0.033) and the ATG16L1 variant (GG vs. AA, OR = 0.4; 95% CI = 0.19–0.81, P = 0.036) were associated with NEC. The Hosmer and Lemeshow Goodness-of-Fit test statistic was P = 0.32.

ATG16L1 variant genotypes and necrotizing enterocolitis (NEC) in the (a) primary and (b) replication cohorts. The proportion of infants who developed NEC stratified by their ATG16L1 (rs2241880) genotypes is shown. The N for NEC infants/all infants within each genotype are as follows: AA- 36/320; AG- 40/478; GG- 10/209. Cochran-Armitage tests were used to evaluate risk associated with the SNP. (a) *P = 0.009 (AA vs. AG vs. GG genotypes; Cochran-Armitage trend test). (b) P = 0.30 (AA vs. AG vs. GG genotypes; Cochran-Armitage trend test).
ATG16L1 Variant in a Replication Cohort
To replicate the association between NEC and the ATG16L1 variant discovered in the primary cohort, we genotyped this variant in an independent replication cohort of 260 premature infants. There were 23 infants with NEC (8.9%) in this replication cohort. Similar to our primary cohort, we found that NEC rates were highest among infants with the AA (12.5%) genotype and lowest among infants with the GG (6.6%) genotype ( Figure 1b ). However, this association was not statistically significant.
ATG16L1 Variant and NEC Outcomes by Race
Because our study cohort was genetically heterogeneous, and genotype frequencies of the ATG16L1 (rs2241880, Thr300Ala) variant differ in African American (AA) and Caucasian (CAU) populations, we examined the relationship between the ATG16L1 SNP and NEC among CAU and AA infants separately. In our primary cohort (n = 1,015), there were 710 CAU infants, 57 (8%) of whom developed NEC. Presence of the A allele was associated with a significant increase (P = 0.02) in NEC rates. Among the 197 AA infants in our primary cohort 23 infants (11.7%) developed NEC. There was a nonsignificant trend (P = 0.10) toward increased NEC among infants with the A allele. To investigate the association between the ATG16L1 variant and NEC in CAU and AA infants in our entire cohort we pooled data from the primary and replication cohorts. The MAF of the G allele was 49% (859/1752) and 34% (191/562) in CAU and AA infants, respectively ( Figure 2 ). NEC was diagnosed in 71/876 (8.1%) CAU infants and 32/281 (11.4%) AA premature infants. The association between the A allele of the ATG16L1 variant and increased NEC was statistically significant in CAU infants (P = 0.01, Cochran Armitage trend) and showed a nonsignificant trend (P = 0.07) in AA infants ( Figure 2 ).

ATG16L1 variant genotypes and necrotizing enterocolitis (NEC) outcomes stratified by race in the combined cohort. NEC outcomes among Caucasian (white bars) and African American (gray bars) infants stratified by ATG16L1 (rs2241880) genotypes is shown. The N for NEC infants/all infants among Caucasian and African American infants were respectively: AA - 27/233; AG - 33/427; GG - 11/216, and AA - 19/125; AG - 11/121; GG - 2/35. Cochran-Armitage tests were used to evaluate risk associated with the SNP. *P = 0.01 (NEC in Caucasians with AA vs. AG vs. GG genotypes): **P = 0.07 (NEC in African Americans with AA vs. AG vs. GG genotypes).
ATG16L1 and Surgical NEC
To examine the relationship between the ATG16L1 variant and disease severity, we used surgical NEC as a marker. In our primary cohort, surgical NEC was diagnosed in 26 infants. In six infants with NEC (n = 86), we did not have information on surgical NEC. There was a statistically nonsignificant trend toward increased surgical NEC with the A allele (AA vs. AG vs. GG; 11/319 (3.5%) vs. 12/475 (2.5%) vs. 3/207 (1.4%)) ( Table 2 ). We found an association between presence of the A allele of the NOD1 (rs6958571) variant and increased NEC (CA trend test; P = 0.042). In regression models that corrected for confounding variables, only GA was associated with surgical NEC (odds ratio: 0.78; 95% CI = 0.65–0.91, P = 0.001).
Discussion
While it is increasingly accepted that genetic factors play a role in NEC pathogenesis, robust susceptibility loci for NEC have not been identified (1,20). Given the importance of the NLR pathway in regulating intestinal immune responses and altering susceptibility to IBD, we postulated that dysregulated NLR signaling will contribute to NEC in premature infants. To test this hypothesis, we took a genetic approach by determining whether common (MAF > 1%), function-altering NLR genetic variants modulate NEC risk in premature infants. The major finding of this study is that a loss of function ATG16lL1 variant (rs2241880, Thr300Ala) was associated with decreased NEC in our primary cohort, and among infants of self-declared Caucasian ancestry. Candidate variants in the NOD1, NOD2, CARD8, and NLRP3 genes were not associated with NEC. Although this study provides preliminary evidence of a role for autophagy in human NEC, lack of correlation with functional data in the preterm intestine and the selective nature of our genotyping approach are limitations of this study.
The ATG16L1 variant (rs2241880, Thr300Ala) has not been investigated with respect to NEC. We found that presence of the A allele at the ATG16L1 locus (g.28152A>G) was associated with increased NEC in our primary cohort. Disease risk increased additively {NEC risk; AA (11%) >AG (7.8%) >GG (3.9%)} with presence of the A allele. The association between the ATG16L1 variant and NEC remained significant after correcting for potential confounders by logistic regression. The minor allele frequency of the G allele among CAU (0.49) and AA (0.34) infants in our study is comparable to the minor allele frequency of the G allele reported in the ExAC browser (http://exac.broadinstitute.org) and published studies (19,23). Similarly, allele frequencies of other variants examined in this study are similar to those reported in prior studies (12,15,24,25). To decrease the possibility of false association arising from analyzing heterogeneous populations we performed race-segregated analysis. The A allele of the ATG16L1 variant was associated with increased NEC in CAU infants, and showed a trend towards increased NEC in AA infants. Interestingly, AA infants who are known to be at higher risk of NEC have a lower frequency of the protective G allele of the ATG16L1 SNP. Although we were not able to replicate our findings in an independent cohort, NEC rates were highest among infants with the AA genotype in the replication cohort. We speculate that the lack of replication is most likely due to a limited sample size of the replication cohort. Similarly, we did not find an association between the ATG16L1 variant and surgical NEC. Possible explanations for these findings are that the variant increases susceptibility to NEC but not severity, center differences in the practice of surgical interventions, presence of other genetic or clinical confounders, and inadequate power to delineate an effect on surgical NEC.
ATG16L1 variants have been extensively studied in Crohn’s disease, a disease characterized by dysregulated NLR responses to gut bacteria (19,23). Presence of the variant G allele at the ATG16L1 locus (g.28152A>G) locus confers additive risk of Crohn’s disease in adults and children (19,23). These results are in contrast with our data that suggest increased risk with the A allele. Potential explanations to explain this discrepancy include developmental state of the immune system in infants vs. adults, and pathophysiological differences between NEC and Crohn’s disease (1,26). NEC develops in the context of immature innate immunity, while Crohn’s disease occurs in the setting of mature intestinal immunity. NEC is postulated to be a phenotype for exaggerated TLR signaling while the pathophysiology of Crohn’s disease involves aberrant NOD-dependent signaling and imbalances in autophagic and adaptive immune responses (1,4,26,27,28). Further, while the microbiome has been implicated in the pathogenesis of both NEC and Crohn’s disease, it evolves dynamically with postnatal maturity in the premature infant (29,30). Intestinal dysbiosis characterized by decreased species diversity, relative abundance of pathogenic bacteria, and an acute increase in Proteobacteria spp. at the expense of Firmicutes spp. has been described in infants who develop NEC (1,29,30). These studies indicate that intestinal dysbiosis creates a proinflammatory milieu in premature infants that can evolve into NEC in susceptible individuals (1,4,6). We speculate that the hypomorphic G allele of ATG16L1 variant may decrease gut dysbiosis-related inflammation, thereby conferring NEC protection. Another key variable that can modulate the gut microbiome, gene–biome interactions and timing of NEC onset is the use of antibiotics in premature infants (1,4,29,30). We did not have adequate data on postnatal age of NEC onset and duration and timing of antibiotic exposure to evaluate the relationships with the ATG16L1 risk allele. Whether autophagy contributes to intestinal adaptation and bacterial immunity in the preterm infant, and whether defective autophagy contributes to NEC pathogenesis remain key questions that need to be investigated.
The mechanism by which the ATG16L1 variant programs aberrant autophagy and inflammation have recently come to light (17,31). ATG16L1 forms a multimeric complex with ATG12 and ATG5, which facilitates elongation and completion of the autophagosome (11,32). NOD1 and NOD2-bacterial sensing recruits ATG16L1 to the plasma membrane with subsequent formation of the autophagosome wrapping bacterial invaders (11,32). The A>G SNP on exon 9 encodes a Thr300 to Ala variant that enhances caspase-mediated protein degradation resulting in decreased function (17,33). In Crohn’s disease, defects in ATG16L1-mediated clearance of bacteria arising from the variant G allele elicits chronic mucosal inflammation and injury (31,33). In contrast, our data indicating less NEC among infants with the variant G allele suggests that decreased autophagic signaling in the developing intestine may be protective. Similar to our results, lack of ATG16L1 has been shown to be protective in the context of intestinal bacterial resistance and carcinoma (16,34). Marchiando et al. (16) showed that ATG16L1-/- mice have a hyperimmune response and less mortality compared to wild-type controls during Citrobacter spp. enteral infections, suggesting that ATG16L1 deficiency confers protection against selective bacteria. In humans with colonic cancer, presence of the variant G allele was associated with increased type I interferon expression, and improved survival (34). These data suggest that decreased autophagy resulting from ATG16L1 variant allele may confer protection against NEC. Studies in animal models suggesting a role for autophagy in propagating NEC support our data demonstrating less NEC with a loss of function autophagy variant (35,36). Access to intestinal tissue from infants with and without NEC would have enabled us to study alterations in intestinal immune and autophagic signaling in relation with the ATG16L1 variant. Investigating whether transgenic mice expressing the mutant allele are more vulnerable to NEC in experimental models (7,28,33) would help clarify the role of ATG16L1 in NEC.
Because NOD1 and NOD2 regulate immune responses to gut bacteria in neonates, we examined candidate variants and NEC outcomes (8,18). The loss of function NOD2 SNP (rs2066844, Arg702Trp) is associated with Crohn’s disease, while the gain of function NOD1 variant (rs6958571), a marker for an indel polymorphism (NOD1 + 32656), has been variably associated with early-onset IBD (8,15,37,38,39,40). The NOD variants were not associated with NEC in our cohort. This is consistent with a prior small study in premature infants showing no association between NOD variants and NEC (41). Similarly, a recent study did not find any association between the NOD2 SNP (rs2066844) and NEC, although presence of two of more NOD2 SNPs was associated with NEC (25). We speculate that while genetically mediated dysregulated NOD signaling plays a role in chronic inflammation characteristic of IBD, it may not contribute to acute inflammation observed in NEC. Two NLRP3 SNPs examined in this study (rs4353135, rs6672995) decrease gene expression, and have been shown to be associated with Crohn’s disease, while the gain of function NLRP3 (rs35829419, p.Q705K) variant has been associated with multiple disease phenotypes in adults (11,13,24,42). CARD8 is known to inhibit Caspase-1 activation and NLRP3-dependent IL-1β activation (43). The CARD8 SNP (rs2043211, p.Cys10X) results in a truncated protein, and has been variably associated with IBD susceptibility (14,26). We did not find an association between NLRP3 or CARD8 variants and NEC. Potential explanations for these negative results could be variation in expression or function of protein in the immature intestine, and preservation of function in the heterozygous state. Given the importance of NOD1, NOD2, and NLRP3 in sensing multiple stress and bacterial signatures, future studies are needed to further examine the role of these genes in NEC.
Previous studies examining the genetic basis of NEC vulnerability have predominantly focused on cytokine and TLR pathway genetic variants (4,5,25,41). Except for Hartel et al. (25), these studies have been generally limited by a small sample size and lack of replication. In this multi-center study, we investigated the relationship between candidate NLR and autophagy SNPs and NEC in a preterm cohort. To the best of our knowledge, this is the first study to examine the relationship between ATG16L1 variants and NEC. We found that a hypomorphic ATG16L1 variant (rs2241880) was less prevalent in infants who developed NEC while functional variants in NOD1, NOD2, and NLRP3 were not associated with NEC. The lack of replication in a smaller cohort, in spite of a similar observable trend limits labeling the ATG16L1 variant as a validated genetic marker. Our results suggest that decreased autophagy resulting from loss of function variants in autophagy genes may protect against NEC. The mechanisms by which ATG16L1 and autophagy regulate host-microbiome interactions in the preterm intestine, and functional implications of the variant examined on these interactions are topics for broader research.
Author Contributions
V.S.: Study design, infant recruitment, data analysis, and manuscript writing. V.B., J.B., P.L., J.G., N.A., N.M., and J.D.: Infant recruitment and manuscript review. D.M. and H.M.: Genotyping and data analysis. L.Z.: Data analysis and manuscript review. Q.M., P.S., and M.D.: Study design, data analysis, and manuscript review.
Statement of Financial Support
Support for this work was provided by Children’s Research Institute of Medical College of Wisconsin (V.S., Milwaukee, WI), KL2TR000056 (CTSI, Southeast Wisconsin), and Children’s Mercy Hospital Research Funds (V.S., Kansas City, MO).
Disclosure
The authors do not have financial or other conflicts to disclose.
References
Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med 2011;364:255–64.
Yee WH, Soraisham AS, Shah VS, Aziz K, Yoon W, Lee SK ; Canadian Neonatal Network. Incidence and timing of presentation of necrotizing enterocolitis in preterm infants. Pediatrics 2012;129:e298–304.
Gordon P, Christensen R, Weitkamp JH, Maheshwari A. Mapping the new world of necrotizing enterocolitis (NEC): review and opinion. EJ Neonatol Res 2012;2:145–72.
Sampath V, Menden H, Helbling D, et al. SIGIRR genetic variants in premature infants with necrotizing enterocolitis. Pediatrics 2015;135:e1530–4.
Treszl A, Tulassay T, Vasarhelyi B. Genetic basis for necrotizing enterocolitis–risk factors and their relations to genetic polymorphisms. Front Biosci 2006;11:570–80.
Afrazi A, Sodhi CP, Richardson W, et al. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr Res 2011;69:183–8.
Sodhi CP, Neal MD, Siggers R, et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012;143:708–18.e1–5.
Chen G, Shaw MH, Kim YG, Nuñez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 2009;4:365–98.
Franchi L, Warner N, Viani K, Nuñez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev 2009;227:106–28.
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 2012;249:158–75.
Carneiro LA, Travassos LH. The interplay between NLRs and autophagy in immunity and inflammation. Front Immunol 2013;4:361.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015;21:677–87.
Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet 2009;41:71–6.
Zhang ZT, Ma XJ, Zong Y, Du XM, Hu JH, Lu GC. Is the CARD8 rs2043211 polymorphism associated with susceptibility to Crohn’s disease? A meta-analysis. Autoimmunity 2015;48:524–31.
Cho JH, Abraham C. Inflammatory bowel disease genetics: Nod2. Annu Rev Med 2007;58:401–16.
Marchiando AM, Ramanan D, Ding Y, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe 2013;14:216–24.
Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014;506:456–62.
Rubino SJ, Selvanantham T, Girardin SE, Philpott DJ. Nod-like receptors in the control of intestinal inflammation. Curr Opin Immunol 2012;24:398–404.
Grigoras CA, Ziakas PD, Jayamani E, Mylonakis E. ATG16L1 and IL23R variants and genetic susceptibility to crohn’s disease: mode of inheritance based on meta-analysis of genetic association studies. Inflamm Bowel Dis 2015;21:768–76.
Bhandari V, Bizzarro MJ, Shetty A, et al.; Neonatal Genetics Study Group. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics 2006;117:1901–6.
Walsh MC, Kliegman RM. Necrotizing enterocolitis: treatment based on staging criteria. Pediatr Clin North Am 1986;33:179–201.
Sampath V, Garland JS, Helbling D, et al. Antioxidant response genes sequence variants and BPD susceptibility in VLBW infants. Pediatr Res 2015;77:477–83.
Salem M, Ammitzboell M, Nys K, Seidelin JB, Nielsen OH. ATG16L1: A multifunctional susceptibility factor in Crohn disease. Autophagy 2015;11:585–94.
Roberts RL, Van Rij AM, Phillips LV, et al. Interaction of the inflammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms. Atherosclerosis 2011;218:123–6.
Härtel C, Hartz A, Pagel J, et al.; German Neonatal Network. NOD2 loss-of-function mutations and risks of necrotizing enterocolitis or focal intestinal perforation in very low-birth-weight infants. Inflamm Bowel Dis 2016;22:249–56.
Stappenbeck TS, Rioux JD, Mizoguchi A, et al. Crohn disease: a current perspective on genetics, autophagy and immunity. Autophagy 2011;7:355–74.
Franchimont D, Vermeire S, El Housni H, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut 2004;53:987–92.
Jilling T, Simon D, Lu J, et al. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol 2006;177:3273–82.
La Rosa PS, Warner BB, Zhou Y, et al. Patterned progression of bacterial populations in the premature infant gut. Proc Natl Acad Sci USA 2014;111:12522–7.
Claud EC, Keegan KP, Brulc JM, et al. Bacterial community structure and functional contributions to emergence of health or necrotizing enterocolitis in preterm infants. Microbiome 2013;1:20.
Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 2010;139:1630–41, 1641.e1–2.
Sorbara MT, Girardin SE. Emerging themes in bacterial autophagy. Curr Opin Microbiol 2015;23:163–70.
Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci USA 2014;111:7741–6.
Grimm WA, Messer JS, Murphy SF, et al. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut 2016;65:456–64.
Yu Y, Shiou SR, Guo Y, et al. Erythropoietin protects epithelial cells from excessive autophagy and apoptosis in experimental neonatal necrotizing enterocolitis. PLoS One 2013;8:e69620.
Neal MD, Sodhi CP, Dyer M, et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J Immunol 2013;190:3541–51.
Alvarez-Lobos M, Arostegui JI, Sans M, et al. Crohn’s disease patients carrying Nod2/CARD15 gene variants have an increased and early need for first surgery due to stricturing disease and higher rate of surgical recurrence. Ann Surg 2005;242:693–700.
van Heel DA, Ghosh S, Butler M, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet 2005;365:1794–6.
Lu WG, Zou YF, Feng XL, et al. Association of NOD1 (CARD4) insertion/deletion polymorphism with susceptibility to IBD: a meta-analysis. World J Gastroenterol 2010;16:4348–56.
Plantinga TS, Fransen J, Knevel R, et al. Role of NOD1 polymorphism in susceptibility and clinical progression of rheumatoid arthritis. Rheumatology (Oxford) 2013;52:806–14.
Szebeni B, Szekeres R, Rusai K, et al. Genetic polymorphisms of CD14, toll-like receptor 4, and caspase-recruitment domain 15 are not associated with necrotizing enterocolitis in very low birth weight infants. J Pediatr Gastroenterol Nutr 2006;42:27–31.
Zhang Q, Fan HW, Zhang JZ, Wang YM, Xing HJ. NLRP3 rs35829419 polymorphism is associated with increased susceptibility to multiple diseases in humans. Genet Mol Res 2015;14:13968–80.
Razmara M, Srinivasula SM, Wang L, et al. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J Biol Chem 2002;277:13952–8.
Acknowledgements
The authors acknowledge the assistance of research nurses Laura Lane RN, BSN (Medical College of Wisconsin), Kathleen Meskin RN, BSN (Medical College of Wisconsin), and Catherine Worwa RN, BSN (Children’s Hospitals and Clinics of Minnesota) for their help with data collection. The authors would like to thank investigators and institutions of the Neonatal Genetics Group: Fernando Moya, MD and Danielle Kurtz, RN, BSN from the New Hanover Regional Medical Center and Coastal Carolina Neonatology, PLLC (Wilmington, NC); Jeffrey Gruen, MD, Matthew Bizzarro, MD, and Haiyeng Meng, MD, PhD from the Yale-New Haven Children’s Hospital and Yale University School of Medicine (New Haven, CT); Claudia Hart, MD from the Magee-Women’s Hospital at UPMC (Pittsburgh, PA); Nirmala S. Desai, MD from the Kentucky Children’s Hospital and University of Kentucky College of Medicine (Lexington, KY); Suhas Nafday, MD from the Montefiore Weiler Hospital and Albert Einstein College of Medicine (Bronx, NY); Manoj Biniwale, MD from the Stony Brook University Medical Center and Stony Brook University School of Medicine (Stony Brook, NY); Juan Sanchez-Esteban, MD from the Women & Infants Hospital of Rhode Island and Warren Alpert Medical School of Brown University (Providence, RI); Vivek Narendran, MD from the Cincinnati Children’s Hospital Medical Center and University of Cincinnati College of Medicine (Cincinnati, OH). Also, thank you to Simran Saini of Temple University for assistance with compilation of the database. The authors would also like to thank all the parents/guardians who consented for their infants to participate in this study.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Sampath, V., Bhandari, V., Berger, J. et al. A functional ATG16L1 (T300A) variant is associated with necrotizing enterocolitis in premature infants. Pediatr Res 81, 582–588 (2017). https://doi.org/10.1038/pr.2016.260
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.260
This article is cited by
-
Anemia, blood transfusions, and necrotizing enterocolitis in premature infants
Pediatric Research (2022)
-
Hope on the horizon: promising novel therapies for necrotizing enterocolitis
Pediatric Research (2020)
-
Intestinal Alkaline Phosphatase Exerts Anti-Inflammatory Effects Against Lipopolysaccharide by Inducing Autophagy
Scientific Reports (2020)
-
New directions in necrotizing enterocolitis with early-stage investigators
Pediatric Research (2020)
-
A miR-511-binding site SNP in the 3′UTR of IGF-1 gene is associated with proliferation and apoptosis of PK-15 cells
In Vitro Cellular & Developmental Biology - Animal (2019)
