
Key Points
-
Alzheimer disease (AD), like other proteinopathic neurodegenerative disorders, is characterized by the accumulation of amyloidogenic proteins
-
A neuroinflammatory component in AD has been known for more than a decade; however, the importance of the contribution of inflammation in the pathogenesis of AD has been appreciated only recently. Genetic and bioinformatic data from individuals with AD and insights from preclinical models now substantiate the present view that inflammation participates in and exacerbates AD pathology.
-
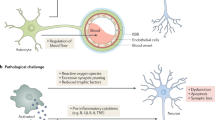
Neuroinflammation in AD is primarily driven by the brain's intrinsic myeloid cells (known as microglia) and escalates with disease progression; thus AD-associated neuroinflammation contrasts with traditionally defined neuroinflammatory diseases such as multiple sclerosis and encephalitides, which are mainly driven by blood-derived leukocytes and B and T lymphocytes, invading the CNS.
-
Manipulation of some of the molecules of the innate immune system or their respective pathways in animal models of AD has resulted in substantial alteration of disease pathology, indicating the potential to ameliorate the disease course through targeting components of the immune system. The immune system thus appears to provide exciting novel and accessible targets for the diagnosis, control and treatment of AD; however, precise knowledge about specific and defined immune events, which may change during the disease course or differ among individuals with AD, is required.
-
Diagnostics research needs to develop sensitive methods to detect immune alterations prior to the onset of AD to identify those patients at risk who may benefit most from specific, tailored anti-inflammatory interventions.
Abstract
The past two decades of research into the pathogenesis of Alzheimer disease (AD) have been driven largely by the amyloid hypothesis; the neuroinflammation that is associated with AD has been assumed to be merely a response to pathophysiological events. However, new data from preclinical and clinical studies have established that immune system-mediated actions in fact contribute to and drive AD pathogenesis. These insights have suggested both novel and well-defined potential therapeutic targets for AD, including microglia and several cytokines. In addition, as inflammation in AD primarily concerns the innate immune system — unlike in 'typical' neuroinflammatory diseases such as multiple sclerosis and encephalitides — the concept of neuroinflammation in AD may need refinement.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Wimo, A. & Prince, M. World Alzheimer Report 2010: The Global Economic Impact of Dementia (Alzheimer's Disease International (ADI), 2010).
Querfurth, H. W. & LaFerla, F. M. Alzheimer's disease. N. Engl. J. Med. 362, 329–344 (2010).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Prokop, S., Miller, K. R. & Heppner, F. L. Microglia actions in Alzheimer's disease. Acta Neuropathol. 126, 461–477 (2013).
Gandy, S. & Heppner, F. L. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron 78, 575–577 (2013).
Perry, V. H. & Holmes, C. Microglial priming in neurodegenerative disease. Nature Rev. Neurol. 10, 217–224 (2014).
Cunningham, C. Microglia and neurodegeneration: the role of systemic inflammation. Glia 61, 71–90 (2013).
Sudduth, T. L., Schmitt, F. A., Nelson, P. T. & Wilcock, D. M. Neuroinflammatory phenotype in early Alzheimer's disease. Neurobiol. Aging 34, 1051–1059 (2013).
Krstic, D. & Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nature Rev. Neurol. 9, 25–34 (2013).
Hickman, S. E. & El Khoury, J. TREM2 and the neuroimmunology of Alzheimer's disease. Biochem. Pharmacol. 88, 495–498 (2014).
Heneka, M. T., Kummer, M. P. & Latz, E. Innate immune activation in neurodegenerative disease. Nature Rev. Immunol. 14, 463–477 (2014).
Akiyama, H. et al. Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421 (2000).
Jonsson, T. et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 (2012).
Scheuner, D. et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature Med. 2, 864–870 (1996). This report showed that all common forms of familial AD could be integrated into a unified pathogenic scheme.
LaFerla, F. M. & Green, K. N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006320 (2012).
Haass, C., Kaether, C., Thinakaran, G. & Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270 (2012).
Holmes, C. et al. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223 (2008).
Giacobini, E. & Gold, G. Alzheimer disease therapy — moving from amyloid-β to tau. Nature Rev. Neurol. 9, 677–686 (2013).
Wyss-Coray, T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nature Med. 12, 1005–1015 (2006).
Zhang, B. et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153, 707–720 (2013). This tour-de-force of bioinformatics illuminated the heretofore cryptic relevance of inflammation-system genes for AD pathogenesis.
Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 (2013).
Guerreiro, R. et al. TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 (2013). These two back-to-back studies identified rare variants of a single myeloid cell receptor as conferring surprisingly high risk for late-onset sporadic AD.
Bradshaw, E. M. et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nature Neurosci. 16, 848–850 (2013).
Tarkowski, E., Andreasen, N., Tarkowski, A. & Blennow, K. Intrathecal inflammation precedes development of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 74, 1200–1205 (2003).
Brosseron, F., Krauthausen, M., Kummer, M. & Heneka, M. T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer's disease: a comparative overview. Mol. Neurobiol. 50, 534–544 (2014).
Krstic, D. et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm. 9, 151 (2012). This study shows that unspecific immune activation maternally and postnatally in wild-type mice can induce full-blown AD pathology, thus causally linking (early) immune stimulation and the development of AD pathology.
Perry, V. H. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 120, 277–286 (2010).
Cunningham, C. et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 65, 304–312 (2009).
Holmes, C. et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774 (2009). This mechanistic clinical research study showed that AD patients with frequent, mild intercurrent infections deteriorated more rapidly, supporting the influence of systemic inflammation on disease course.
Cribbs, D. H. et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J. Neuroinflamm. 9, 179 (2012).
Schwartz, M. & Shechter, R. Systemic inflammatory cells fight off neurodegenerative disease. Nature Rev. Neurol. 6, 405–410 (2010).
Kyrkanides, S. et al. Osteoarthritis accelerates and exacerbates Alzheimer's disease pathology in mice. J. Neuroinflamm. 8, 112 (2011).
Abuabara, K. et al. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the U.K. Br. J. Dermatol. 163, 586–592 (2010).
Gisondi, P. et al. Mild cognitive impairment in patients with moderate to severe chronic plaque psoriasis. Dermatology 228, 78–85 (2014).
Thaler, J. P. et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 122, 153–162 (2012).
Takeda, S. et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl Acad. Sci. USA 107, 7036–7041 (2010).
Mayeux, R. et al. Genetic susceptibility and head injury as risk factors for Alzheimer's disease among community-dwelling elderly persons and their first-degree relatives. Ann. Neurol. 33, 494–501 (1993).
Heneka, M. T. et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 26, 1343–1354 (2006).
Rubartelli, A. & Lotze, M. T. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28, 429–436 (2007).
Bornemann, K. D. et al. Aβ-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am. J. Pathol. 158, 63–73 (2001).
Stalder, A. K. et al. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J. Neurosci. 25, 11125–11132 (2005).
Eikelenboom, P. et al. Neuroinflammation in Alzheimer's disease and prion disease. Glia 40, 232–239 (2002).
Streit, W. J. Microglia and Alzheimer's disease pathogenesis. J. Neurosci. Res. 77, 1–8 (2004).
Prinz, M., Priller, J., Sisodia, S. S. & Ransohoff, R. M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nature Neurosci. 14, 1227–1235 (2011).
International Multiple Sclerosis Genetics Consortium et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nature Genet. 43, 436–441 (2011).
Thambisetty, M. et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol. Psychiatry 73, 422–428 (2013).
Lambert, J. C. et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nature Genet. 41, 1094–1099 (2009).
Hollingworth, P. et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature Genet. 43, 429–435 (2011).
Liang, Y. & Tedder, T. F. Identification of a CD20-, FcɛRIβ-, and HTm4-related gene family: sixteen new MS4A family members expressed in human and mouse. Genomics 72, 119–127 (2001).
Ransohoff, R. M. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nature Neurosci. 15, 1074–1077 (2012).
Raj, T. et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014).
Iwashyna, T. J., Ely, E. W., Smith, D. M. & Langa, K. M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304, 1787–1794 (2010).
Prinz, M. & Priller, J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nature Rev. Neurosci. 15, 300–312 (2014).
Du Yan, S. et al. Amyloid-β peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc. Natl Acad. Sci. USA 94, 5296–5301 (1997).
El Khoury, J. et al. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 382, 716–719 (1996).
Bamberger, M. E., Harris, M. E., McDonald, D. R., Husemann, J. & Landreth, G. E. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J. Neurosci. 23, 2665–2674 (2003).
Paresce, D. M., Ghosh, R. N. & Maxfield, F. R. Microglial cells internalize aggregates of the Alzheimer's disease amyloid β-protein via a scavenger receptor. Neuron 17, 553–565 (1996).
Stewart, C. R. et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nature Immunol. 11, 155–161 (2010).
Sheedy, F. J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nature Immunol. 14, 812–820 (2013).
Koenigsknecht, J. & Landreth, G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846 (2004).
Fassbender, K. et al. The LPS receptor (CD14) links innate immunity with Alzheimer's disease. FASEB J. 18, 203–205 (2004).
El Khoury, J. B. et al. CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 197, 1657–1666 (2003).
Griffin, W. S. et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl Acad. Sci. USA 86, 7611–7615 (1989).
Patel, N. S. et al. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J. Neuroinflamm. 2, 9 (2005).
Vom Berg, J. et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nature Med. 18, 1812–1819 (2012). This study showed that genetic as well as pharmacological blocking of the IL-12–IL-23 pathway substantially ameliorated AD pathology in an AD mouse model and provided the first hints that IL-12 and IL-23 are upregulted in the CSF of patients with AD, thus offering a druggable immune target made for repurposing existing IL-12 and IL-23 inhibitors.
Fillit, H. et al. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci. Lett. 129, 318–320 (1991).
Tan, J. et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nature Neurosci. 5, 1288–1293 (2002).
Tan, J. et al. Microglial activation resulting from CD40–CD40L interaction after β-amyloid stimulation. Science 286, 2352–2355 (1999).
Jin, J. J., Kim, H. D., Maxwell, J. A., Li, L. & Fukuchi, K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J. Neuroinflamm. 5, 23 (2008).
Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nature Rev. Mol. Cell Biol. 8, 101–112 (2007).
Lee, C. Y. & Landreth, G. E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960 (2010).
Streit, W. J., Sammons, N. W., Kuhns, A. J. & Sparks, D. L. Dystrophic microglia in the aging human brain. Glia 45, 208–212 (2004).
Perry, V. H., Nicoll, J. A. & Holmes, C. Microglia in neurodegenerative disease. Nature Rev. Neurol. 6, 193–201 (2010).
Krabbe, G. et al. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8, e60921 (2013).
Hickman, S. E., Allison, E. K. & El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28, 8354–8360 (2008).
Lucin, K. M. et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 79, 873–886 (2013). This paper shows that beclin 1 is altered in microglia derived from AD patients and provides the first molecular explanations of some aspects of microglial impairment in AD.
Mawuenyega, K. G. et al. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science 330, 1774 (2010).
Grathwohl, S. A. et al. Formation and maintenance of Alzheimer's disease β-amyloid plaques in the absence of microglia. Nature Neurosci. 12, 1361–1363 (2009).
Parkhurst, C. N. et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). This paper demonstrates that microglia have an important role in learning and memory by providing neurotrophic factors, such as BDNF.
Hong, S. et al. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319 (2014).
Selkoe, D. J. SnapShot: pathobiology of Alzheimer's disease. Cell 154, 468–468 e1 (2013).
Verdier, Y., Zarandi, M. & Penke, B. Amyloid β–peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease. J. Pept. Sci. 10, 229–248 (2004).
Wake, H., Moorhouse, A. J. & Nabekura, J. Functions of microglia in the central nervous system — beyond the immune response. Neuron Glia Biol. 7, 47–53 (2011).
Salter, M. W. & Beggs, S. Sublime microglia: expanding roles for the guardians of the CNS. Cell 158, 15–24 (2014).
Frank, S. et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447 (2008).
Hickman, S. E. et al. The microglial sensome revealed by direct RNA sequencing. Nature Neurosci. 16, 1896–1905 (2013).
Hsieh, C. L. et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156 (2009).
Kleinberger, G. et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl Med. 6, 243ra86 (2014).
Wang, Y. et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 (2015).
Jay, T. R. et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 (2015). These two publications show the importance and function of TREM2 in AD mouse models; that is, to promote the survival of activated microglia and myeloid cells, to recruit these cells to A β plaques through sensing for A β -associated lipids, and to modulate hippocampal A β burden.
Ulrich, J. D. et al. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 9, 20 (2014).
Lajaunias, F., Dayer, J. M. & Chizzolini, C. Constitutive repressor activity of CD33 on human monocytes requires sialic acid recognition and phosphoinositide 3-kinase-mediated intracellular signaling. Eur. J. Immunol. 35, 243–251 (2005).
Griciuc, A. et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643 (2013).
Miller, K. R. & Streit, W. J. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 3, 245–253 (2007).
El Khoury, J. et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature Med. 13, 432–438 (2007).
Hawkes, C. A. & McLaurin, J. Selective targeting of perivascular macrophages for clearance of β-amyloid in cerebral amyloid angiopathy. Proc. Natl Acad. Sci. USA 106, 1261–1266 (2009).
Hickey, W. F. & Kimura, H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239, 290–292 (1988).
Lai, A. Y. & McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: the issue of what, when and where. Future Neurol. 7, 165–176 (2012).
Mildner, A. et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J. Neurosci. 31, 11159–11171 (2011).
Gomez-Nicola, D., Schetters, S. T. & Perry, V. H. Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 62, 1041–1052 (2014).
Burda, J. E. & Sofroniew, M. V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248 (2014).
Sofroniew, M. V. & Vinters, H. V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 (2010).
Medeiros, R. & LaFerla, F. M. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 239, 133–138 (2013).
Heneka, M. T. et al. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflamm. 2, 22 (2005).
Olabarria, M., Noristani, H. N., Verkhratsky, A. & Rodriguez, J. J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer's disease mouse model: mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 6, 55 (2011).
Olabarria, M., Noristani, H. N., Verkhratsky, A. & Rodriguez, J. J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia 58, 831–838 (2010).
Furman, J. L. et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease. J. Neurosci. 32, 16129–16140 (2012).
Nagele, R. G., D'Andrea, M. R., Lee, H., Venkataraman, V. & Wang, H. Y. Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209 (2003).
Wyss-Coray, T. et al. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nature Med. 9, 453–457 (2003). This paper demonstrates that astrocytes can have an important impact on catabolising A β.
Koistinaho, M. et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nature Med. 10, 719–726 (2004).
Terwel, D. et al. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J. Neurosci. 31, 7049–7059 (2011).
Pihlaja, R. et al. Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59, 1643–1657 (2011).
Saido, T. & Leissring, M. A. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2, a006379 (2012).
Wyss-Coray, T. & Rogers, J. Inflammation in Alzheimer disease — a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2, a006346 (2012).
Roth, A. D., Ramirez, G., Alarcon, R. & Von Bernhardi, R. Oligodendrocytes damage in Alzheimer's disease: beta amyloid toxicity and inflammation. Biol. Res. 38, 381–387 (2005).
Mitew, S. et al. Focal demyelination in Alzheimer's disease and transgenic mouse models. Acta Neuropathol. 119, 567–577 (2010).
Hosokawa, M., Klegeris, A., Maguire, J. & McGeer, P. L. Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia 42, 417–423 (2003).
Yamada, T., Akiyama, H. & McGeer, P. L. Complement-activated oligodendroglia: a new pathogenic entity identified by immunostaining with antibodies to human complement proteins C3d and C4d. Neurosci. Lett. 112, 161–166 (1990).
Harrison, J. K. et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl Acad. Sci. USA 95, 10896–10901 (1998).
Lee, S. et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am. J. Pathol. 177, 2549–2562 (2010).
Singhrao, S. K., Muller, C. T., Gilbert, S. J., Duance, V. C. & Archer, C. W. An immunofluorescence method for postembedded tissue in the acrylic resin Technovit 9100 New using fluorescein isothiocyanate secondary detection. Microsc. Res. Tech. 72, 501–506 (2009).
Yang, L. B., Li, R., Meri, S., Rogers, J. & Shen, Y. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer's disease. J. Neurosci. 20, 7505–7509 (2000).
Walker, D. G., Dalsing-Hernandez, J. E., Campbell, N. A. & Lue, L. F. Decreased expression of CD200 and CD200 receptor in Alzheimer's disease: a potential mechanism leading to chronic inflammation. Exp. Neurol. 215, 5–19 (2009).
Sagare, A. P., Bell, R. D. & Zlokovic, B. V. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a011452 (2012).
Vukic, V. et al. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer's brain is mediated by the JNK–AP1 signaling pathway. Neurobiol. Dis. 34, 95–106 (2009).
Palmer, J. C., Barker, R., Kehoe, P. G. & Love, S. Endothelin-1 is elevated in Alzheimer's disease and upregulated by amyloid-β. J. Alzheimers Dis. 29, 853–861 (2012).
Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J. Neuroinflamm. 8, 26 (2011).
Lyros, E., Bakogiannis, C., Liu, Y. & Fassbender, K. Molecular links between endothelial dysfunction and neurodegeneration in Alzheimer's disease. Curr. Alzheimer Res. 11, 18–26 (2014).
Chiang, K. & Koo, E. H. Emerging therapeutics for Alzheimer's disease. Annu. Rev. Pharmacol. Toxicol. 54, 381–405 (2014).
Michelucci, A., Heurtaux, T., Grandbarbe, L., Morga, E. & Heuschling, P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-β. J. Neuroimmunol. 210, 3–12 (2009).
Martinez, F. O. & Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13 (2014).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Anand, P., Singh, B., Jaggi, A. S. & Singh, N. Mast cells: an expanding pathophysiological role from allergy to other disorders. Naunyn Schmiedebergs Arch. Pharmacol. 385, 657–670 (2012).
Piette, F. et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer's disease: a randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 3, 16 (2011).
Breitner, J. C. & Zandi, P. P. Do nonsteroidal antiinflammatory drugs reduce the risk of Alzheimer's disease? N. Engl. J. Med. 345, 1567–1568 (2001).
in t' Veld, B. A. et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med. 345, 1515–1521 (2001).
Aisen, P. S. The potential of anti-inflammatory drugs for the treatment of Alzheimer's disease. Lancet Neurol. 1, 279–284 (2002).
Group, A. R. et al. Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 65, 896–905 (2008).
Group, A. R. et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808 (2007).
Aisen, P. S. et al. Effects of rofecoxib or naproxen versus placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826 (2003).
Breitner, J. C. et al. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 7, 402–411 (2011).
Lo, A. W., Ho, C., Cummings, J. & Kosik, K. S. Parallel discovery of Alzheimer's therapeutics. Sci. Transl Med. 6, 241cm5 (2014).
Guerreiro, R. J. et al. Peripheral inflammatory cytokines as biomarkers in Alzheimer's disease and mild cognitive impairment. Neurodegener. Dis. 4, 406–412 (2007).
Hu, W. T. et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 79, 897–905 (2012). Using an unbiased proteome approach, this study provides evidence for changes in immune-relevant factors in the plasma of MCI and AD patients.
Liu, Y. et al. Interleukin-23 receptor polymorphisms are associated with Alzheimer's disease in Han Chinese. J. Neuroimmunol. 271, 43–48 (2014).
Tan, M. S. et al. IL12/23 p40 inhibition ameliorates Alzheimer's disease-associated neuropathology and spatial memory in SAMP8 mice. J. Alzheimers Dis. 38, 633–646 (2014).
Griffin, W. S. Neuroinflammatory cytokine signaling and Alzheimer's disease. N. Engl. J. Med. 368, 770–771 (2013).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nature Immunol. 9, 857–865 (2008).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013). This study shows that NLRP3 activation occurs in microglia in patients with AD and provides evidence that inhibition of NLRP3 reduces AD pathology in vivo.
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Med. 21, 248–255 (2015).
Yamanaka, M. et al. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331 (2012).
Cramer, P. E. et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506 (2012).
Veeraraghavalu, K. et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-f (2013).
Tesseur, I. et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-e (2013).
Price, A. R. et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-d (2013).
Fitz, N. F., Cronican, A. A., Lefterov, I. & Koldamova, R. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924-c (2013).
Fuhrmann, M. et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nature Neurosci. 13, 411–413 (2010).
Nash, K. R. et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 34, 1540–1548 (2013).
Frenkel, D. et al. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nature Commun. 4, 2030 (2013).
Wyss-Coray, T. et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc. Natl Acad. Sci. USA 99, 10837–10842 (2002).
Fonseca, M. I., Zhou, J., Botto, M. & Tenner, A. J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J. Neurosci. 24, 6457–6465 (2004).
Maier, M. et al. Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J. Neurosci. 28, 6333–6341 (2008).
Paolicelli, R. C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011).
Schafer, D. P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Stephan, A. H., Barres, B. A. & Stevens, B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389 (2012).
van der Wal, E. A., Gómez-Pinilla, F. & Cotman, C. W. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 4, 69–72 (1993).
Wyss-Coray, T. et al. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nature Med. 7, 612–618 (2001).
Town, T. et al. Blocking TGF-β–Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nature Med. 14, 681–687 (2008). This paper demonstrates that inhibition of TGF β in myeloid cells can reduce AD pathology in mice.
Swardfager, W. et al. A meta-analysis of cytokines in Alzheimer's disease. Biol. Psychiatry 68, 930–941 (2010).
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P. & Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49, 489–502 (2006).
Bard, F. et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of alzheimer disease. Nature Med. 6, 916–919 (2000).
Garcia-Alloza, M. et al. A limited role for microglia in antibody mediated plaque clearance in APP mice. Neurobiol. Dis. 28, 286–292 (2007).
Golde, T. E., Das, P. & Levites, Y. Quantitative and mechanistic studies of Aβ immunotherapy. CNS Neurol. Disord. Drug Targets 8, 31–49 (2009).
Koenigsknecht-Talboo, J. et al. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci. 28, 14156–14164 (2008).
Wilcock, D. M. et al. Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J. Neurosci. 23, 3745–3751 (2003).
Wilcock, D. M. et al. Microglial activation facilitates Aβ plaque removal following intracranial anti-Aβ antibody administration. Neurobiol. Dis. 15, 11–20 (2004).
Wilcock, D. M. et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. 24, 6144–6151 (2004).
Wang, A., Das, P., Switzer, R. C. 3rd, Golde, T. E. & Jankowsky, J. L. Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-β immunotherapy. J. Neurosci. 31, 4124–4136 (2011).
Citron, M. Alzheimer's disease: strategies for disease modification. Nature Rev. Drug Discov. 9, 387–398 (2010).
Medeiros, R. et al. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am. J. Pathol. 182, 1780–1789 (2013).
Kiyota, T. et al. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther. 19, 724–733 (2012).
Chakrabarty, P. et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 85, 519–533 (2015).
Jin, P. et al. Anti-inflammatory and anti-amyloidogenic effects of a small molecule, 2,4-bis(p-hydroxyphenyl)-2-butenal in Tg2576 Alzheimer's disease mice model. J. Neuroinflamm. 10, 2 (2013).
Chakrabarty, P. et al. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol. Neurodegener. 7, 36 (2012).
Imbimbo, B. P. et al. CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer's disease. Br. J. Pharmacol. 156, 982–993 (2009).
Sivilia, S. et al. Multi-target action of the novel anti-Alzheimer compound CHF5074: in vivo study of long term treatment in Tg2576 mice. BMC Neurosci. 14, 44 (2013).
CERESPIR. CERESPIR Incorporated is pleased with positive interim Phase 2 results for CHF 5074 in patients with mild cognitive impairment, presented by Chiesi at the AAIC 2013 Meeting in Boston. CERESPIR [online], (2013).
Ross, J. et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: a 12-week, double-blind, placebo-controlled study. Curr. Alzheimer Res. 10, 742–753 (2013).
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Villeda, S. A. et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nature Med. 20, 659–663 (2014). These two studies show that changes in blood-borne factors including immune molecules such as the chemokine CCL11 are linked to impaired neurogenesis and decline in cognitive performance during ageing, which can be rescued by transfer of young blood to aged mice.
Mackaness, G. B. Cellular resistance to infection. J. Exp. Med. 116, 381–406 (1962).
Nathan, C. F., Murray, H. W., Wiebe, M. E. & Rubin, B. Y. Identification of interferon-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 158, 670–689 (1983).
Stein, M., Keshav, S., Harris, N. & Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292 (1992).
Mosmann, T. R., Cherwinski, H., Bond, M. W., Giedlin, M. A. & Coffman, R. L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357 (1986).
Mills, C. D., Kincaid, K., Alt, J. M., Heilman, M. J. & Hill, A. M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–6173 (2000).
Vahedi, G., Kanno, Y., Sartorelli, V. & O'Shea, J. J. Transcription factors and CD4 T cells seeking identity: masters, minions, setters and spikers. Immunology 139, 294–298 (2013).
Hirahara, K. et al. Helper T-cell differentiation and plasticity: insights from epigenetics. Immunology 134, 235–245 (2011).
Josefowicz, S. Z. Regulators of chromatin state and transcription in CD4 T-cell polarization. Immunology 139, 299–308 (2013).
Lawrence, T. & Natoli, G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nature Rev. Immunol. 11, 750–761 (2011).
Lacey, D. C. et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J. Immunol. 188, 5752–5765 (2012).
Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014).
Chiu, I. M. et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401 (2013).
Mucke, L. & Selkoe, D. J. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338 (2012).
Cui, Y. H. et al. Up-regulation of FPR2, a chemotactic receptor for amyloid β 1–42 (Aβ42), in murine microglial cells by TNFα. Neurobiol. Dis. 10, 366–377 (2002).
Lotz, M. et al. Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J. Neurochem. 94, 289–298 (2005).
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G. & Landreth, G. E. CD14 and Toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 29, 11982–11992 (2009).
Hu, J., Akama, K. T., Krafft, G. A., Chromy, B. A. & Van Eldik, L. J. Amyloid-β peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 785, 195–206 (1998).
Larson, M. et al. The complex PrPc–Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. J. Neurosci. 32, 16857-71a (2012).
Papassotiropoulos, A. et al. A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer's disease. Ann. Neurol. 45, 666–668 (1999).
Chakrabarty, P. et al. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 24, 548–559 (2010).
Chakrabarty, P., Herring, A., Ceballos-Diaz, C., Das, P. & Golde, T. E. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 6, 16 (2011).
Li, R. et al. Tumor necrosis factor death receptor signaling cascade is required for amyloid-β protein-induced neuron death. J. Neurosci. 24, 1760–1771 (2004).
Cheng, X., Yang, L., He, P., Li, R. & Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer's disease and non-demented patients. J. Alzheimers Dis. 19, 621–630 (2010).
Tobinick, E. L. & Gross, H. Rapid cognitive improvement in Alzheimer's disease following perispinal etanercept administration. J. Neuroinflamm. 5, 2 (2008).
Ryu, J. K. & McLarnon, J. G. Block of purinergic P2X7 receptor is neuroprotective in an animal model of Alzheimer's disease. Neuroreport 19, 1715–1719 (2008).
Diaz-Hernandez, J. I. et al. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer's disease through GSK3β and secretases. Neurobiol. Aging 33, 1816–1828 (2012).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB TRR 43, NeuroCure Exc 257 and HE 3130/6-1 to FLH), the Federal Ministry of Education and Research (DLR/BMBF; Kompetenznetz Degenerative Demenzen), the European Union ITN-NeuroKine project, and a Collaborative Research Grant of the Berlin Institute of Health (BIH) to F.L.H. Work in the Ransohoff lab has been supported by the U. S. National Institutes of Health, the National Multiple Sclerosis Society, the Williams Family Fund for MS Research and the Guthy Jackson Charitable Foundation. Work in the Becher lab is supported by grants from the Swiss National Science Foundation (316030_150768, 310030_146130 and CRSII3_136203), European Union FP7 project TargetBraIn, NeuroKine and ATECT, and the university research priority project translational cancer research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
F.L.H. and B.B. hold a patent application entitled 'Modulators of IL-12 and/or IL-23 for the Prevention or Treatment of Alzheimer's Disease' (PCT/EP2012/050066) and are founding scientists of Myosotis Therapeutics AG, which has exclusive licensing rights from the University of Zurich and the Charité –Universitätsmedizin Berlin. R.M.R. is an employee of Biogen.
Glossary
- Myeloid cells
-
The subset of leukocytes that are not lymphocytes. They include granulocytes, monocytes, macrophages and dendritic cells.
- Familial AD
-
An uncommon form of AD that usually occurs before the age of 65 and is inherited in an autosomal dominant fashion.
- Microglia activation
-
A term used to describe a functional activation of microglial cells, for example, in response to a defined stimulus in pathophysiological settings or during development; however, it is often used to describe a change in the morphological appearance of microglia that does not necessarily correspond to the functional status of these cells. Reactive microglia is a term used when microglia respond to pathological changes and deviate from the normal steady-state.
- Encephalitides
-
Acute inflammatory diseases of the brain, typically consisting of tissue-invading leukocytes (mainly T cells).
- Quantitative trait loci
-
Stretches of DNA that contain or are linked to the genes that underlie a quantitative trait. Quantitative traits refer to certain phenotypes.
- Cerebral amyloidosis
-
This term describes all forms of CNS diseases that feature the deposition of proteins (so-called proteinopathies).
- Hypertrophy
-
The increase in the volume of an organ, tissue or cell.
- Apolipoprotein E
-
(APOE). A class of apolipoprotein that is required for the catabolism of triglyceride-rich lipoprotein constituents. In the CNS, APOE is generated primarily by astrocytes, and transports cholesterol to neurons via APOE receptors, whereas in the periphery, APOE is mainly produced by the liver and macrophages, and mediates cholesterol metabolism.
- Neurovascular unit
-
(NVU). This consists of vascular cells such as brain endothelial cells, pericytes and vascular smooth muscle cells, glial cells such as astrocytes, microglia and oligodendroglia, and neurons. It links neural activity to blood flow and controls the exchange of biologically relevant protein interactions between brain and the periphery.
- Adaptive immune system
-
The immune system that forms the basis for acquired immunity and immunological memory and involves T and B lymphocytes.
- Mast cells
-
These are resident granulocytes of several types of tissues containing many granules rich in histamine and heparin.
- Biologicals
-
Medicinal products that are manufactured in or extracted from biological sources (they may also be termed biopharmaceutical or biologic medical products); they are distinct from chemically synthesized pharmaceutical products.
- NLRP3 inflammasome
-
Inflammasomes comprise a sensor molecule from the NOD-like receptor (NLR) family or the pyrin and HIN domain-containing protein (PYHIN) family, the adaptor protein ASC and caspase 1. The NALP3 inflammasome is expressed in myeloid cells, senses a wide range of aggregated molecules, and promotes the maturation of the inflammatory cytokines interleukin-1 (IL-1) and IL-18.
Rights and permissions
About this article

Cite this article
Heppner, F., Ransohoff, R. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372 (2015). https://doi.org/10.1038/nrn3880
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrn3880
This article is cited by
-
iPSC-derived PSEN2 (N141I) astrocytes and microglia exhibit a primed inflammatory phenotype
Journal of Neuroinflammation (2024)
-
Identification of immunogenic cell death-related genes involved in Alzheimer’s disease
Scientific Reports (2024)
-
The HLA-DRB1*09:01-DQB1*03:03 haplotype is associated with the risk for late-onset Alzheimer’s disease in APOE \({{\varepsilon }}\)4–negative Japanese adults
npj Aging (2024)
-
Early modulation of the gut microbiome by female sex hormones alters amyloid pathology and microglial function
Scientific Reports (2024)
-
Reduced insulin/IGF-1 signalling upregulates two anti-viral immune pathways, decreases viral load and increases survival under viral infection in C. elegans
GeroScience (2024)
