
Key Points
-
Pancreatic cancer evolves in three stages: initiation, expansion and survival in foreign microenvironments.
-
Factors that contribute to initiation are inherited (germline) mutations, somatic mutations acquired during organ growth and renewal, ageing, chronic inflammation, obesity and smoking.
-
Genetic mutations in KRAS, cyclin-dependent kinase inhibitor 2A (CDKN2A), TP53 and SMAD4 drive clonal expansion by conferring a selective growth advantage on pancreatic cancer cells.
-
Selection pressures and bottlenecks result from extension into new microenvironments of immune cells, stroma, organ-specific cell types and resource gradients that vary spatially and temporally. Metastasis requires dispersal, invasion and colonization of microenvironments distant from the pancreatic primary site.
-
Remaining questions include the order of early cancer-promoting events, the phenotypic importance of late-occurring mutations and the clinical relevance of genetic, microenvironmental and cellular heterogeneity.
-
Evolutionary thinking provides a framework for the biology of pancreatic cancer, revealing how and why this lethal tumour evolves.
Abstract
Cancer is an evolutionary disease, containing the hallmarks of an asexually reproducing unicellular organism subject to evolutionary paradigms. Pancreatic ductal adenocarcinoma (hereafter referred to as pancreatic cancer) is a particularly robust example of this phenomenon. Genomic features indicate that pancreatic cancer cells are selected for fitness advantages when encountering the geographic and resource-depleted constraints of the microenvironment. Phenotypic adaptations to these pressures help disseminated cells to survive in secondary sites, a major clinical problem for patients with this disease. In this Review we gather the wide-ranging aspects of pancreatic cancer research into a single concept rooted in Darwinian evolution, with the goal of identifying novel insights and opportunities for study.
Similar content being viewed by others

Main
The question is not what you look at, but what you see. Henry David Thoreau1
In the year 2016, an estimated 53,070 patients will be diagnosed with pancreatic ductal adenocarcinoma (hereafter referred to as pancreatic cancer), most of whom will die of their disease within 5 years2. There are no clinically validated screening methods for pancreatic cancer in the curative stage, and surgery remains the only option for cure, despite the fact that only 10–15% of newly diagnosed patients are deemed eligible3. Few other effective treatment modalities exist that significantly extend overall survival4. Ultimately, most patients will die with metastases to the liver, lung and/or peritoneum, the most common sites of spread5. Patients, clinicians and researchers alike are frustrated at the lack of progress being made, indicating that new strategies are needed to understand this disease.
The term 'cancer' engenders fear and anger, particularly when one is newly faced with the devastating diagnosis of pancreatic cancer. Moreover, a common reaction is to personify the cancer as an evil entity that must be battled to save the patient's life. The weapons for this battle include a surgeon's scalpel, chemotherapy, radiation, targeted agents, holistic approaches and religious faith. But, in a biological sense what really is a pancreatic cancer, or any cancer (Box 1)? Once the above- mentioned preconceived biases are removed, pancreatic cancer reveals itself as a robust example of Darwinian evolution, a pervasive phenomenon in the natural world that is subject to its own rules, restraints and predictable characteristics. Cancer has been discussed in evolutionary terms for 40 years, first by Peter Nowell6 in 1976, who proposed clonal evolution as a unifying model of tumour initiation and progression based on his observations in haematopoietic malignancies. However, the importance of evolutionary dynamics for understanding cancer was brought to the forefront by the application of next-generation sequencing methodologies to cancer samples7. This has certainly been the case for pancreatic cancer, in which recurrent chromosome abnormalities and subclonal events were described by karyotypic analysis almost two decades ago8, whereas the description of intratumoural heterogeneity based on next-generation sequencing was reported only in 2010 (Refs 9,10).
Three evolutionary stages
An understanding of pancreatic cancer in evolutionary terms is perhaps best accomplished by characterizing it into three broad stages11. These are initiation of the tumour by the acquisition of a driver gene mutation in a cell of origin, clonal expansion of the mutation-carrying cell into a multicellular neoplasm and the introduction of the neoplastic cells into both local and distant microenvironments.
Initiation
A basic tenet of Darwinian evolution is that purposeless mutations occur randomly in asexually reproducing cells upon which selection then acts12,13. For a mutation to occur, a complete cell division must take place. Likewise, the occurrence of a somatic mutation implies that at least one cell division occurred in the lineage that gave rise to that cell12. Given that the expected somatic mutation rate is approximately three single nucleotide variants per cell division14 and that the adult pancreas is not a highly proliferative tissue15, by simple chance alone it is exceedingly rare for an initiating driver gene mutation to occur (Fig. 1a). In patients who develop sporadic pancreatic cancer, the appearance of the driver gene mutation in the first cell is predicted to occur at least two decades before diagnosis9.

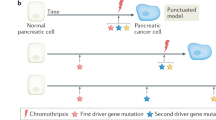
a | Stage 1: initiation. A normal cell of the pancreas acquires an initiating driver gene mutation as a result of environmental exposure or a lapse in DNA repair. In most instances, this initiating mutation causes the cell to undergo apoptosis or senescence, or to be lost owing to immune surveillance or during a bottleneck event or tissue turnover (genetic drift). If these mechanisms fail, the cell carrying the initiating mutation (red) escapes from senescence and immunosuppression, and continues to fixation because of a survival or growth advantage. b | Stage 2: clonal expansion. The cell carrying the initiating mutation and its progeny continue to divide, creating a clonal population defined by the presence of the driver gene mutation. In the stepwise progression model, as the population grows in both cell number and geographic space, the descendants gradually acquire additional driver gene mutations (dark beige, blue and green cells in the left panel) and passenger mutations that increase clonal heterogeneity of the neoplasm. In the punctuated evolution model, a catastrophic genome-wide event occurs in a single cell cycle that results in widespread structural damage and acquisition of multiple driver gene alterations simultaneously (green cells in the right panel). c | Stage 3: introduction to foreign microenvironments. Ongoing clonal expansion may lead to a population of cells (green cells) that break through the basement membrane into the surrounding stroma. This event represents a genetic bottleneck that leads to a reduction in genetic diversity. Additional genetic events, signals provided by the stroma, deposition of dense extracellular matrix (ECM) and immune infiltrates all provide selective forces that shape the adaption of these cells into subclonal populations that differ with respect to their overall fitness (represented by cells coloured different shades of green). Dissemination is probably an ongoing process during tumour development; however, the extent to which cells from the entire neoplasm uniformly enter the circulation and/or whether dissemination is restricted to a subpopulation is unknown. Nonetheless, those disseminated cells that achieved high fitness in the primary site may have the greatest chance of colonizing new microenvironments, such as the liver, lung or peritoneum, common sites of metastasis in pancreatic cancer. Colonization of secondary sites represents yet another genetic bottleneck that may further reduce genetic heterogeneity. CDKN2A, cyclin-dependent kinase inhibitor 2A; MDSC, myeloid-derived suppressor cell; WT, wild type.
Unlike proliferative tissues such as the breast or colon, in which familial cancers occur 10–20 years earlier than sporadic cancers, inheritance of a high-risk variant for developing pancreatic cancer decreases its latency by only 5 years16, underscoring that the number of cell divisions and time are important factors in the initiation of pancreatic cancer. Recently, a statistical analysis of numerous cancer types, including pancreatic cancer, determined a strong correlation of lifetime risk with the number of normal stem cell divisions in a tissue17. On the basis of estimates of normal pancreatic stem cell renewal rates18,19, stochastic mistakes during DNA replication (intrinsic factors) were predicted to substantially contribute to the lifetime risk of pancreatic cancer. However, the relative contribution of intrinsic versus extrinsic factors in cancer initiation has stimulated vigorous scientific debate, with a follow-up study20 concluding that the influence of extrinsic factors such as radiation and carcinogens far outweighs that of intrinsic factors. Nonetheless the study by Wu et al.20 also showed that nearly half of pancreatic cancer mutations were probably caused by intrinsic factors.
Recent whole-genome sequencing of 593 patients with familial pancreatic cancer indicates that the genetic basis of familial pancreatic cancer is polygenic, that is, many kindreds had one or more high-risk germline variants, but the frequency of any one variant never exceeded 3% of the population studied21. The best-studied germline variants linked to pancreatic cancer risk are BRCA1, BRCA2, partner and localizer of BRCA2 (PALB2), the Fanconi anaemia genes FANCC and FANCG, and ataxia telangiectasia mutated (ATM), which are all components of the DNA double-strand break repair machinery16,22. Mutations in these genes (specifically, BRCA1 and BRCA2) increase genomic instability during faulty homologous recombination at stalled replication forks and hence increase the rate at which somatic mutations occur23. Germline mutations in cyclin-dependent kinase (CDK) inhibitor 2A (CDKN2A, which encodes p16INK4A and p19ARF), responsible for familial atypical multiple mole melanoma syndrome, are also strongly associated with an increased risk of pancreatic cancer and melanoma16, presumably through loss of the G1/S checkpoint. Mutations in DNA double-strand break repair genes probably increase the rate at which the initial driver gene mutation occurs per cell division, consistent with the concept that these are caretaker genes. However, the tumour suppressive function of p16INK4A indicates that it is a gatekeeper gene24. Thus, its loss would be predicted to increase the number of cell divisions, increasing the chance that additional driver gene mutations could occur.
Epidemiological studies point to several risk factors for developing pancreatic cancer that also fit into this framework25. For example, patients with chronic pancreatitis owing to protease, serine 1 (PRSS1) or serine peptidase inhibitor, Kazal type 1 (SPINK1) mutations have a well-documented increased risk of developing pancreatic cancer, perhaps as a result of the increased epithelial cell divisions that occur during injury and repair processes, or from reactive oxygen species that cause DNA damage26. Both the ongoing cycles of injury and repair and reactive oxygen species would be expected to increase the net number of mutations that occur per division. Inflammatory processes may further enable clonal populations to survive and expand that otherwise would be removed by the immune system27. Likewise, smoking contributes mutagens that cause DNA damage in pancreatic cells, thereby promoting the initiating event, clonal expansion and the accumulation of additional mutations as well25,28. Obesity is thought to increase pancreatic cancer risk by inducing a chronic pro-inflammatory state and hyperinsulinaemia25,29. Type II diabetes, another well-known association with pancreatic cancer, is also thought to increase risk through hyperinsulinaemia and/or the hyperglycaemia caused by the dysregulation of blood glucose levels30. Hyperglycaemia may increase cancer risk by supporting the survival and expansion of KRAS mutant clones that have differential dependence on glucose metabolism31. Finally, genome-wide association studies have identified various susceptibility loci for pancreatic cancer predicted to modify the net rates of cell growth (telomerase reverse transcriptase (TERT), nuclear receptor subfamily 5 group A member 2 (NR5A2), zinc and ring finger 3 (ZNRF3) and TP63 (Refs 32,33,34)) or efficiency of DNA repair (structural maintenance of chromosomes 2 (SMC2))34.
Clonal expansion
The occurrence of the initiating driver gene mutation does not guarantee the development of pancreatic cancer, as the mutation must become fixed in the epithelial cell population (Fig. 1a). Up to 33% of pancreata from autopsy series contain pancreatic intraepithelial neoplasias (PanINs), known precursor lesions of pancreatic cancer (Box 2), buttressing the notion that most PanINs never progress to an infiltrating carcinoma35.
The extent to which the nascent tumour cell then undergoes additional cell divisions enabling the gradual accumulation of somatic alterations over time (stepwise progression, also known as linear progression) or rapidly over a limited number of cell cycles (punctuated) is unknown (Fig. 1b). Support for the stepwise progression model stems from classic evidence demonstrating increasing frequency of KRAS, CDKN2A, TP53 (which encodes p53) and SMAD4 alterations with increasing atypia of PanINs36. High-sensitivity methods to detect KRAS mutations indicate that they are present in more than 99% of stage 1 PanIN (PanIN-1) lesions. Moreover, although KRAS mutations are present in all PanIN stages, the proportion of cells containing KRAS mutations increases with PanIN grade, supporting the finding that this population expands clonally37. Loss of p16INK4A protein expression can be demonstrated in PanIN-2 and PanIN-3, with the frequency of p16INK4A loss higher in PanIN-3 (Ref. 38); similarly, TP53 nuclear accumulation or SMAD4 loss has been demonstrated in PanIN-3 and invasive cancers, and the frequency of somatic alteration of both genes is higher in invasive cancer39,40. Such patterns indicate waves of clonal expansion in association with the accumulation of driver gene alterations. By contrast, punctuated evolution, or chromothripsis, is defined as the acquisition of numerous structural alterations in a single cell cycle by a catastrophic genomic event41. Evidence of chromothripsis has been found in 10% of pancreatic cancers42. Although that study did not characterize the genetic alterations caused by chromothripsis specifically, there was no evidence that chromothripsis was a dominant mechanism of driver gene alteration in those cancers. Thus, although it is entirely plausible that chromothripsis can disrupt driver genes in a stochastic manner, upon which positive selection can act, punctuated evolution does not seem to be as common a pathway as stepwise progression in pancreatic carcinogenesis.
Irrespective of how they accumulate, the genetic landscape of pancreatic cancer is dominated by three or four mountains represented by somatic alterations in KRAS, CDKN2A, TP53 and SMAD4 (Refs 42,43,44,45), all of which have been shown to arise in PanINs36,46. Advances in sequencing technology and throughput and increasing sample sizes have not altered this terrain, suggesting that the discovery phase of high-frequency genetic targets in pancreatic cancer has reached saturation. It also indicates that there are few evolutionary paths to the formation of pancreatic cancer. However, such efforts continue to be fruitful in identifying previously unknown low-frequency events of significance42,44,45. Recurrent somatic alterations are perhaps best understood in the context of the pathway or function affected, for many cases of low-frequency targets reveal themselves to be alternative perturbations of a common pathway43 (Table 1).
KRAS. Activating mutations of the KRAS oncogene on chromosome 12p are the most common genetic abnormality, present in approximately 95% of pancreatic tumours analysed44,47. KRAS encodes a member of the RAS family of GTP-binding proteins that mediate many cellular functions, including proliferation, cell survival and cytoskeletal remodelling48. Most mutations in KRAS are believed to cause a constitutively active protein, although recently some KRAS mutants, specifically KRASG12C, have been demonstrated to have nucleotide cycling activity that is druggable49,50. In approximately 4% of pancreatic cancers KRAS amplification occurs together with the oncogenic mutation42. BRAF, the signalling mediator immediately downstream of KRAS, is mutated or amplified in a mutually exclusive manner from KRAS in 3–4% of cases42,51. Intriguingly, although KRAS mutations are found in 99% of PanIN-1s37, no more than 95% of pancreatic cancers have a KRAS or BRAF mutation, supporting the notion that a KRAS mutation is not strictly required for the development of pancreatic cancer.
CDKN2A. The tumour suppressor gene CDKN2A encodes p16INK4A and p19ARF through a common locus on chromosome 9p52. The genomic structure of CDKN2A is highly complex in that it produces two mRNAs, each with a unique first exon but sharing exons 2 and 3. However, exon 2 in the mRNA that encodes p19ARF is derived from a different reading frame from that of the mRNA encoding p16INK4A; thus, the two proteins are not isoforms53. The high frequency at which this locus is inactivated in pancreatic cancers (>90%)54 raises the question of which tumour suppressor is being selected for inactivation55. Evidence in mice and humans points to p16INK4A as the primary target because mutations in exon 1 — which is used in the transcript encoding p16INK4A — that would leave p19ARF functional have been reported in both pancreatic cancers and melanomas55. However, large homozygous deletions often inactivate both proteins so that loss of either may contribute to pancreatic carcinogenesis by different mechanisms. For example, p16INK4A inhibits cell cycle progression through the G1/S checkpoint mediated by CDKs such as CDK4 and CDK6 (Ref. 56). The loss of this important restraint leads to unchecked CDK4 and CDK6 expression and cell cycle progression through the G1/S checkpoint. Telomere shortening in concert with the loss of the G1/S checkpoint creates an environment that facilitates chromosome instability and the accumulation of structural rearrangements, including fold-back inversions, which are a form of mutation relatively specific to pancreatic cancer10,57. By contrast, p19ARF induces cell-cycle arrest independently of CDKs by binding to the E3 ubiquitin ligase MDM2 to inhibit p53 degradation; loss of p19ARF abrogates p53-induced apoptosis and cell cycle arrest55. In a small number of pancreatic cancers that retain CDKN2A, somatic mutations in F-box and WD repeat domain containing 7 (FBXW7), which encodes a ubiquitin ligase that targets cyclin E for degradation, or the gene encoding the ring E3 ubiquitin ligase anaphase promoting complex subunit 2 (APC2, also known as ANAPC2) that regulates chromosome segregation and mitotic fidelity have been reported43,51.
TP53. p53 is a latent transcription factor that is activated by stimuli such as DNA damage or stress. Upon activation, p53 performs many functions, including regulation of the G1/S checkpoint, maintenance of G2/M arrest to enable DNA repair, and apoptosis58. TP53 is somatically mutated in up to 85% of pancreatic cancers47. As many as 66% of TP53 mutations in pancreatic cancer are missense mutations that affect the DNA binding domain43,47. Although not completely inactivating, such mutations provide oncogenic gains of function compared with the normal protein58, often in association with nuclear accumulation of p53 in the neoplastic cell39. Previous studies based on immunolabelling for the p53 protein product in fixed tissues have drastically underestimated the frequency with which TP53 is inactivated by not accounting for somatic alterations that result in a loss of protein expression39,59. However, it is clear that nonsense mutations, frameshifts and homozygous deletions not detected by p53 immunolabelling are notable mechanisms of TP53 inactivation in pancreatic cancer43,44. In one study of late-stage pancreatic cancers, up to half of all mutations in TP53 were predicted to cause a loss of protein expression leading to null alleles47. Among cancers that have no detectable TP53 mutation, numerous genes, such as excision repair cross-complementation group 4 (ERCC4), ERCC6, E1A binding protein p300 (EP300) or RAN binding protein 2 (RANBP2), are mutated at low frequencies and may provide alternative inactivation of one or more p53 functions. Of particular importance is ATM, a gene also implicated in familial pancreatic cancer that may also be sporadically mutated60; ATM phosphorylates p53 directly and has pivotal roles in responding to cell stress and maintaining genome integrity61.
SMAD4. SMAD4 is inactivated in approximately 55% of pancreatic cancers, either by homozygous deletion (30%) or by an intragenic mutation in association with loss of the second copy (25%)62. The SMAD4 protein is a crucial co-transcription factor and mediator of the transforming growth factor-β (TGFβ) canonical signalling pathway for cellular growth, differentiation and maintenance of tissue homeostasis63. The TGFβ pathway is notable for its dualistic nature in cancer; during the early stages of the clonal expansion phase (PanIN-1 and PanIN-2) it restrains neoplastic cell growth, whereas in later stages of clonal expansion (PanIN-3 and invasive cancers) TGFβ signalling promotes growth, in part owing to the loss of SMAD4 and the canonical arm of the TGFβ pathway64,65. Up to 10% of pancreatic cancers without SMAD4 alterations harbour an inactivating mutation in TGFβ receptor 1 (TGFBR1), TGFBR2, activin A receptor type 1B (ACVR1B) or SMAD3, providing alternative mechanisms to inactivate TGFβ signalling43,44.
An important facet of SMAD4 inactivation is the context in which it occurs. One study that evaluated the patterns of coexistence of driver gene mutations found that most pancreatic cancers with SMAD4 inactivation had coexistent TP53 gain-of-function alterations, whereas pancreatic cancers that retained SMAD4 were more likely to have TP53 loss-of-function alterations47. This relationship probably reflects the interdependence of p53 and TGFβ for transcriptional gene activation (discussed in greater detail in the next section). Thus, TP53 mutant pancreatic cancers can be segregated into two types, those with TP53 loss of function and wild-type SMAD4, and those with TP53 gain of function and SMAD4 loss of function. Validation of these genetic subtypes in independent cohorts and their relationship to therapeutic responses remain to be discerned, including the extent to which these genetic subtypes overlap with other biological subtypes that have been described66,67,68 (Box 3). Such efforts would be particularly worthwhile in the context of clinical trials of patients with pancreatic cancer for which pretreatment tissues are available69.
Synergistic effects between mutations. Although the genes described thus far reveal those pathways that are disrupted, it is unlikely that each gene exerts its effects independently. Mutant KRAS has been shown to impede TGFβ signalling by inhibiting receptor SMADs70,71 and inhibiting p53 by blocking its amino-terminal phosphorylation72. In turn, TGFβ signalling interacts at many levels with the RAS–RAF–ERK pathway73. p53 is required for TGFβ target gene transactivation by binding to distinct cis-enhancer elements in the same gene promoters through association with receptor SMADs74. Furthermore, mutant p53 and SMADs form a complex that inhibits p63, enabling aggressive features of cancer cells75. Thus, disruption of crucial driver genes creates a complex tumorigenic network that is expected to greatly alter the systems biology of the cell. An improved understanding of this altered system could be exploited to identify unique vulnerabilities and target specific mutant proteins or pathways76.

Introduction to foreign microenvironments
Extension of the neoplastic clonal population from the ductal system into the adjacent pancreatic stroma parallels the introduction of a species into a novel environment77,78 (Fig. 1c). Invasion into the novel environment is in no way guaranteed however, as the clonal population probably requires a threshold number of genetic, epigenetic and phenotypic alterations to successfully invade and colonize. This concept is exemplified by mouse PanIN-2 and PanIN-3 lesions from which cells may disseminate and survive in the liver but do not persist to form secondary masses79. Whether this moment of invasion represents the passive displacement of neoplastic epithelial cells through an incompetent basement membrane, positive selection by one or more microenvironmental factors, or both, is unknown. Regardless, the microenvironment of a primary pancreatic cancer comprises various cell types, extracellular matrix (ECM) components and restricted nutrient and oxygen gradients80,81,82,83,84 that act as potent selection forces and shape the ongoing adaptation and continued clonal expansion of this parental clone. In turn, the phenotypes of the neoplastic cells undergo random modifications, one or more of which might support cell survival and maximize fitness in that microenvironment at that moment in time. The result is a primary tumour that is heterogeneous at the cell autonomous and non-cell autonomous levels. Excellent reviews of the pancreatic cancer microenvironment already exist80,81,82,83,84; thus, in this section we will focus only on how they relate to evolutionary paradigms.
The desmoplastic stroma of pancreatic cancer. The epithelial wound healing response is particularly robust in the pancreas, as evidenced by histology findings in pancreata from patients with chronic pancreatitis85. This response is evolutionarily conserved to support the multicellular state and is coordinated in large part by TGFβ (Ref. 86). Features associated with a wound healing response include fibroblast activation, immune suppression, remodelling of the ECM and trophic signals to promote re-epithelialization87. That the stroma en masse has an influence on the neoplastic epithelium is undisputed, yet the extent to which each cell type supports rather than restrains neoplastic growth is an area of immense interest.
Co-option of the stromal response by cancer indicates that the stroma provides paracrine signals that select tumour cells with certain properties. Such paracrine signals originate from various sources but have mostly been described for α-smooth muscle actin (αSMA)+ myofibroblasts. Myofibroblasts are derived from normally quiescent pancreatic stellate cells (PSCs) in the pancreas. Upon activation, PSCs lose their cytoplasmic lipid, transdifferentiate into αSMA+ myofibroblasts with proliferative capacity, secrete various growth factors, such as TGFβ, fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF), and substantially increase production of ECM components88. Moreover, unlike the non-neoplastic setting, in which proliferative signals are eventually quelled with the culmination of repair following injury, PSCs and other stromal mesenchymal cells are continuously activated by the neoplastic epithelium itself, which secretes PDGF, TGFβ and sonic hedgehog (SHH)89,90,91. Evidence supporting the tumour-promoting role of PSCs and their myofibroblastic derivatives comes from mouse studies in which pharmacological inhibition of PSC activation by the vitamin D analogue calcipotriol led to stromal collapse, smaller tumours and improved chemotherapeutic delivery92. Analogous results are seen following stromal ablation by short-term inhibition of Hedgehog signalling93 or enzymatic ablation of hyaluronic acid (HA), a major constituent of the ECM that is secreted by myofibroblasts94,95. By contrast, two recent studies using a mouse model of pancreatic cancer indicated that stromal ablation by conditional deletion of Shh (chronic inhibition)96 in or of αSMA+ myofibroblasts themselves97 led to more aggressive tumours. This suggests that distinct components of the myofibroblastic secretome have tumour-restraining properties, although one cannot entirely rule out that the secretomes of other stromal cell populations (for example, macrophages) have tumour suppressive features as well. These opposing forces occur over geographic space and time and in part may underlie the formation of intratumoural heterogeneity by favouring selective sweeps of one clonal population at the expense of another.
The effects of the microenvironment on pancreatic cancer cells go beyond stromal cells. The abundant ECM produced by αSMA+ myofibroblasts is rich in HA, fibrillar collagens and secreted protein acidic and rich in cysteine (SPARC), which act as a physical barrier to the neoplasm94,95. HA is a large negatively charged glycosaminoglycan that binds to large amounts of water, leading to high hydrostatic pressure and interstitial fluid pressure (IFP)80. The swelling caused by high concentrations of HA stresses collagen fibrils tethered to cancer or endothelial cell surface receptors, which contract in response, leading to pathological IFP, widespread vascular collapse and hypoperfusion. Although this is problematic from the point of view of therapeutic delivery93,94, such a phenomenon itself may cause geographic isolation of neoplastic cells that are already in a nutrient-restricted environment, thus enforcing allopatric evolution and intratumoural heterogeneity. IFP also leads to hypoxia, a pervasive feature of the pancreatic cancer microenvironment that serves as yet another powerful selective force93. Pancreatic cancer cells can adapt to these environmental pressures through metabolic reprogramming and shunting of resources; these adaptations occur in association with KRAS mutations well before the onset of invasion and are continuously refined with subclonal evolution83,98. This is analogous to ecological studies that have shown that the most successful invasive species are those that are predisposed to the most efficient use of available (limited) resources78.
The immune system in pancreatic cancer. The immune system represents yet another highly complex programme that has evolved to support the multicellular state99. In the context of cancer evolution immune cells represent native predators. Abundant evidence indicates that the pancreatic cancer microenvironment is immunosuppressed at multiple levels, some of which occur in association with the clonal expansion phase of the neoplasm and themselves may enforce genetic bottlenecks in a temporal and spatial manner100. In general, the pancreatic cancer microenvironment is notable for T cell suppression by several mechanisms, including an accumulation of CD4+ forkhead box P3 (FOXP3)+ regulatory T cells (Treg cells), M2 tumour-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) and fibroblast activation protein (FAP)+ fibroblasts, a type of stromal cell distinct from αSMA+ myofibroblasts in the pancreatic cancer microenvironment84,101,102. The endogenous cytotoxic T cells do not seem to be dysfunctional, as mechanisms to bypass their suppression unmask latent immune responses and promote intratumoural accumulation of T cells101,103.
Metastasis of pancreatic cancer. Although metastasis is managed clinically as a distinct stage, from an evolutionary standpoint it is a reflection of clonal competition and fitness levels in the primary site (Fig. 1c). Four lines of evidence support this notion. First, dissemination itself has not been shown to be a rate-limiting step for the formation of metastases, as 99% of cells that enter the circulation do not survive beyond 24 hours104; moreover, dissemination occurs from the earliest stages of pancreatic carcinogenesis79. Second, no metastasis-specific genes have been found in pancreatic cancer; instead, a substantial proportion of metastatic efficiency is determined by the genetic alterations that arise during the clonal expansion phase itself (KRAS, CDKN2A, TP53 and SMAD4), before the moment of invasion47,65. Thus, the genetic features of the parental clone play an important part (albeit they are not the only factor) in determining the extent to which the clone will successfully adapt and survive in foreign microenvironments should metastasis occur. Third, in two independent studies encompassing 13 unique patients, metastatic subclones were shown to arise from large populations of cells in the primary tumour10,105. These subclones can be identified by their unique set of passenger mutations or structural rearrangements, which are genetic markers of the life history of that lineage9. Finally, mathematical models predict at least 5–10 years for the emergence of a metastatic subclone following development of the parental clone, again implying the importance of time for adaptation within the microenvironment9. The complement of features of the pancreatic cancer cell or its microenvironment that dictate metastasis to the liver, lungs or peritoneum has yet to be determined. However, recent data using lineage tracing in a mouse model of pancreatic cancer indicate that multiclonal seeding is required to initiate metastases in an organ-specific manner105.
Unanswered questions
Is KRAS the only initiating driver gene in sporadic pancreatic cancer? KRAS is undoubtedly important for pancreatic cancer biology, and extensive efforts are under way to target this oncoprotein106. What is debatable is whether KRAS mutations are the only initiating event in pancreatic cancer.
Oncogenic KRAS mutations can be found in the pancreata of patients with no evidence of PanINs or pancreatic cancer, suggesting that they are necessary but not sufficient to initiate pancreatic carcinogenesis107. Moreover, people with germline mutations in KRAS have not been reported to have a higher risk of developing pancreatic cancer or other non-neoplastic pancreatic sequelae; instead they develop diseases related to developmental delay, bone marrow failure and syndromic cardio-facio-cutaneous disorders, all probably a result of oncogene-induced senescence108. Thus, it seems paradoxical that KRAS mutations that may cause senescence can initiate pancreatic cancer. One explanation for this paradox is that the spectrum of KRAS germline mutations differs with respect to the codons affected and thus they do not lead to KRAS hyperactivity to the same extent as oncogenic mutations, for example, G12S, K117R and A146T mutations, in patients with Costello syndrome compared with G12D mutations in those with pancreatic cancers108.
Compelling experimental evidence that supports the notion that mutant KRAS in preductal epithelial cells can initiate pancreatic cancer is its ability to inhibit immune-induced senescence and promote localized immunosuppression27,109,110. Oncogenic KRAS has also been shown to induce expression of functional interleukin-17 (IL-17) receptors on transformed epithelial cells while stimulating infiltration of IL-17-producing T helper 17 (TH17) cells and γδ T cells into the adjacent microenvironment. As a result, the transformed epithelial cells undergo paracrine stimulation by the secreted IL-17, which supports clonal expansion of the KRAS mutant population111. Thus, although the random occurrence of an oncogenic KRAS mutation may cause senescence in most instances, occasionally the KRAS mutant cell may survive long enough to incite a local immunotolerant environment that supports its clonal expansion into a large enough population for additional genetic events to occur112 (Fig. 1a). This scenario is consistent with mathematical models that predict that at least a decade is required from initiation to formation of the clonal population that will eventually breach the basement membrane and become an infiltrating carcinoma9.
An alternative possibility that should be considered is that mutations in KRAS are not always the initiating event but may be a driver gene alteration that is selected for in the clonal expansion phase following a tumour cell of origin first acquiring a different driver gene mutation. An example of a strong candidate for an alternative initiating driver gene is CDKN2A, as CDKN2A mutations are linked to an inherited risk of pancreatic cancer21. Inherited mutations in DNA damage repair genes such as BRCA2 do not negate the possibility that genes such as CDKN2A could be an alternative initiating driver, as they act by increasing the number of potentially deleterious genetic events per cell division and hence the chance that inactivating mutations in these driver genes occur. Consistent with this interpretation, there is no difference in the genetics of familial pancreatic cancers compared with those in which the disease occurred sporadically21. Finally, although not experimentally studied, constitutional epimutation may be a mechanism of pancreatic carcinogenesis113. This would be supported by reports that many patients with a strong familial pattern of inheritance do not have an identifiable germline genetic event21. It is crucial to understand these possibilities in light of limited success in screening for pancreatic cancer or in developing chemopreventive strategies thus far. For example, anti-inflammatory or immunomodulatory agents may have a greater preventive effect for KRAS-initiated pancreatic cancers than for those that arise as a result of loss of a tumour suppressive mechanism114.
What is the importance of mutations in epigenome regulatory genes? Perhaps the biggest revelation from high-throughput sequencing of many cancer types, including pancreatic cancer, has been the identification of recurrent somatic mutations in genes encoding epigenome regulators, specifically members of the SWI/SNF complex and the histone-lysine N-methyltransferase 2 (KMT2) family. Individually, members of each gene family are somatically mutated in a small percentage (<5%) of pancreatic cancers analysed42,43,44,45. Collectively, the frequency of somatic alterations for any member of these gene families is higher, suggesting that a common epigenomic phenotype is selected for by various genotypes. For example, up to 30% of pancreatic cancers were shown to have an alteration in one of five different members of the SWI/SNF complex in a mutually exclusive manner115.
The SWI/SNF family of genes encodes proteins that make up one of two complexes, the BRG1- or HRBM-associated factor (BAF) complex and the polybromo-associated BAF (PBAF) complex. Each complex relies on ATP hydrolysis to directly disrupt histone–DNA contacts. In general the key role for SWI/SNF complexes is to control the balance between differentiation and stemness and to antagonize the action of the Polycomb repressive complex116. Although mutations in several SWI/SNF family members have been described in pancreatic cancer, the most frequently mutated genes are AT-rich interactive domain-containing 1A (ARID1A) and SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2 (SMARCA2), both components of the BAF complex42,44. Similar to genes related to SWI/SNF signalling, the KMT2 genes encode proteins that are components of multi-subunit complexes. KMT2 proteins methylate histone H3 on lysine 4 (H3K4) to promote genome accessibility and transcription117. In pancreatic cancer, KMT2C (also known as MLL3) is the most commonly mutated member of this family, although mutations in KMT2D (also known as MLL2) and KMT2A (also known as MLL) are also seen42,44. Like SWI/SNF, KMT2 genes have pervasive roles in regulating stemness and differentiation.
To date, the temporal occurrence of these gene alterations has not been explored, thus it remains to be seen whether they represent mutations acquired during the clonal expansion phase or subclonal events that are selected for their fitness advantage in the primary tumour microenvironment. This distinction is crucial, as events acquired during early carcinogenesis are expected to be more targetable than those that are subclonal in nature11. In addition, unlike CDKN2A, TP53 or SMAD4, in which both alleles are targeted, members of the SWI/SNF and KMT2 gene families require loss of function of only a single allele for their effects in cancer116,117. A determination of the requirement of the wild-type allele for cancer cell survival would be fruitful, as it may provide a therapeutic vulnerability using synthetic-lethal approaches118,119. The temporal occurrence of these alterations also has importance from the evolutionary perspective. Mutations that arise during the intraductal clonal expansion phase provide a clue to the survival advantage required for the neoplasm to develop, and suggest that at the moment of invasion the parental clone was already maximally equipped for survival through rapid epigenetic adaptation. By contrast, mutations that arise in a subclonal manner after invasion occurs may be a reflection of the spatial heterogeneity of microenvironmental selection factors. Such an instance could be exploited to better understand the heterogeneity of the microenvironment in general and in relation to stromal ablation therapies (Fig. 2).

Geographic heterogeneity refers to the spatial variation within a single patient's tumour with respect to genotypes and phenotypes. a | Spatial concordance. In spatial concordance, genetic subclonal populations and regions of distinct stromal biology, immune and/or metabolic phenotypes are geographically linked, as shown in the serial sections of a hypothetical primary tumour. This pattern would be consistent with genotypic heterogeneity driving phenotypic heterogeneity within a neoplasm. A representative example of a genotype that directly drives phenotypic features in pancreatic cancer are KRAS mutations that lead to immunological and metabolic alterations27,142. In this scenario, targeting of subclones or the dominant subclone that drives tumour progression may be most efficacious. b | Spatial discordance. In spatial discordance, genotypic and phenotypic variations are unrelated to each other, implying that phenotypic variations in distinct regions of a tumour are unrelated to genotype and are more influenced by epigenomic or polygenic models of tumour behaviour. Unlike targeting of subclones, in this situation methods to modulate the epigenome to reduce cellular plasticity may have greater value. Spatial discordance has not been formally shown in human tumours because so far all global analyses have relied on single tumour samples, and thus it is of theoretical concern only until proved.
What are the clinically relevant aspects of heterogeneity? Heterogeneity is a loosely used term in cancer biology. At one extreme it may be used to describe inter-patient heterogeneity with respect to biological subtypes of the disease that differ in their aetiology21, biology66,67,68 or response to therapy42 (Box 3). The success of any personalized intervention depends on the specific genotype and microenvironment, and the immune and metabolic phenotypes of each patient. There is no doubt that a better understanding of such phenotypes will provide rapid improvements in clinical management, as it has already in BRCA-mutant ovarian cancers120. At the opposite extreme is intratumoural heterogeneity, most often described in relation to genetics9, although epigenetic or phenotypic variants of pancreatic cancer can be described with this term121.
Broadly in the field of cancer research there is a lack of distinction between genetic subclonal heterogeneity within a primary tumour in general, in metastasis- initiating cells of the primary tumour specifically, or within a metastasis, each of which may have distinct clinical and therapeutic implications at a particular stage of disease122 (Fig. 3). A deeper understanding of these different types of heterogeneity will help to define clinically relevant subclones, and the contexts in which subclonal heterogeneity is most meaningful biologically and therapeutically (Fig. 2). Moreover, there is little distinction between heterogeneity of unequivocal driver gene alterations and heterogeneity of somatic alterations with predicted consequences in passenger genes. However, the latter provide unexplored territory with regard to the importance of spatially distinct passenger mutations within a single neoplasm in relation to immunoediting123, the mini-driver model of polygenic cancer evolution124 or recurrent regions of haploinsufficiency125. A counterintuitive view also asks to what extent is heterogeneity reduced during tumour evolution? Although mutations and cell divisions supply heterogeneity over time, there probably exist several bottleneck events that reduce overall diversity during cancer evolution, for example, fixation in evolutionary stage 1 (initiation) and colonization in evolutionary stage 3 (introduction to foreign microenvironments).

a | Intratumoural heterogeneity within a primary tumour. The founder clone (indicated by the grey cell) is the ancestral cell population whose lineage contains all mutations acquired post-fertilization by the most recent common ancestor in the primary tumour. Thus, each mutation that was present in the founder cell is present in every descendant subclone and is inferred by the trunk of the phylogeny that contains n mutations. The founder cell itself no longer exists, as once it divides and accumulates a new mutation or mutations it has evolved. Subclone 1 (red cells) is composed of cells that have acquired mutation a. Subclone 2 (blue cells) and subclone 3 (beige cells) are also descendants of the founding cell that have acquired mutations d and b, respectively. Subclone 4 (green cells) has mutations b and c, indicating that it shares a common ancestor with subclone 3. b | Intratumoural heterogeneity within a metastasis. The metastasis-initiating cell (dark green cell) contains the initial, distinct set of mutations common to all cells of the metastasis (genotype n plus mutations b and c from panel a). The metastasis- initiating cell itself no longer exists, as once it divides and accumulates a new mutation or mutations it has evolved. Subclone 1 (light green cell) represents direct descendants of the metastasis-initiating cell that acquired mutation x, and subclone 2 (pink cell) represents the direct descendants that acquired mutation y. Subclone 3 (dark beige cell) has mutations y and z, indicating that it shares a common ancestor with subclone 2. c | Intratumoural heterogeneity of metastasis-initiating cells within a primary tumour. The metastasis-initiating cells share a common ancestor, yet, nonetheless, have distinct mutations that distinguish one from the other (that is, blue versus green genotypes). As each initiating cell is the ancestral cell for its respective metastasis, every descendant cell will inherit this founding set of mutations.
What are the evolutionary effects of treatment? Currently, the only potentially curative therapy for pancreatic cancer is surgical removal of the neoplasm3, causing an evolutionary effect akin to near total decimation of the cancer cell population. However, most patients who undergo surgery will develop recurrent disease, providing evidence that small populations of cancer cells are left behind either locoregionally or systemically and as predicted by computational models126. The evolutionary dynamics by which these residual cells survive, divide and develop into clinically evident populations of cancer cells while under the selective pressures of systemic chemotherapy or radiation is unknown.
The same can be stated for locally advanced, unresectable or metastatic pancreatic cancer. It is reasonable to assume that radiation, cytotoxic chemotherapies and targeted agents that constitute the standards of care for this disease all influence cancer cell evolution13. However, at each stage of disease the extent to which different treatment modalities contribute to genetic bottlenecks, the selection of resistant clones or the de novo formation of resistant clones remains unknown despite our general knowledge of resistance mechanisms in cancer127. Only with dedicated studies that rely on post-treatment tissues at the time of progression can these questions begin to be addressed.
Summary
The pace of discovery in understanding pancreatic cancer biology is at its height. Compared with less than one decade ago we have a firm grasp of the genome of pancreatic cancer43,44 and the mechanisms by which metabolism is altered to suit the needs of pancreatic cancer cells83, and insight into rationally targeting the nodes of immunosuppression102 or exploiting genomic instability42. However, what is lacking is a convergence of these parallel lines of study, as they are no doubt highly interrelated. This Review has attempted to collate the current understanding of pancreatic cancer into a single concept rooted in evolutionary biology. Mechanisms to support cross-collaboration of these exciting areas of research are expected to further accelerate the pace of discovery and ultimately improve patient survival.
References
Bradford Torrey, E. The Writings of Henry David Thoreau (Riverside Press, 1906).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2016. CA Cancer J Clin. 66, 7–30 (2016).
Winter, J. M. et al. Survival after resection of pancreatic adenocarcinoma: results from a single institution over three decades. Ann. Surg. Oncol. 19, 169–175 (2012).
Castellanos, E., Berlin, J. & Cardin, D. B. Current treatment options for pancreatic carcinoma. Curr. Oncol. Rep. 13, 195–205 (2011).
Yachida, S. & Iacobuzio-Donahue, C. A. The pathology and genetics of metastatic pancreatic cancer. Arch. Pathol. Lab. Med. 133, 413–422 (2009).
Nowell, P. C. The clonal evolution of tumor cell populations. Science 194, 23 (1976). Landmark perspective that enumerated the roles of genetic variation, natural selection and evolution in the progression of cancer. Also highlighted how these processes may confound therapeutics.
Brosnan, J. A. & Iacobuzio-Donahue, C. A. A new branch on the tree: next-generation sequencing in the study of cancer evolution. Semin. Cell Dev. Biol. 23, 237–242 (2012).
Gorunova, L. et al. Cytogenetic analysis of pancreatic carcinomas: intratumor heterogeneity and nonrandom pattern of chromosome aberrations. Genes Chromosomes Cancer 23, 81–99 (1998). One of the first studies to catalogue intratumoural karyotypic heterogeneity in pancreatic cancer.
Yachida, S. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010). Demonstrated that pancreatic cancer metastases evolve from geographic primary tumour subclones. Also modelled the required time for tumour evolution, suggesting that pancreatic cancer takes many years to develop metastases.
Campbell, P. J. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113 (2010). Used phylogenetic approaches to interpret structural rearrangements and the relationship of intrapatient metastases for a set of patients with pancreatic cancer. Also found that genomic instability continues during tumour evolution.
Vogelstein, B. & Kinzler, K. W. The path to cancer —three strikes and you're out. N. Engl. J. Med. 373, 1895–1898 (2015).
Altrock, P. M., Liu, L. L. & Michor, F. The mathematics of cancer: integrating quantitative models. Nat. Rev. Cancer 15, 730–745 (2015).
Greaves, M. & Maley, C. C. Clonal evolution in cancer. Nature 481, 306–313 (2012). Modern synthesis of tumour evolutionary concepts and Darwinian selection in cancer.
Bozic, I. et al. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl Acad. Sci. USA 107, 18545–18550 (2010). First to quantify the survival growth advantage conferred by a driver gene mutation.
Klein, W. M., Hruban, R. H., Klein-Szanto, A. J. P. & Wilentz, R. E. Direct correlation between proliferative activity and dysplasia in pancreatic intraepithelial neoplasia (PanIN): additional evidence for a recently proposed model of progression. Mod. Pathol. 15, 441–447 (2002).
Petersen, G. M. Familial pancreatic adenocarcinoma. Hematol. Oncol. Clin. North Am. 29, 641–653 (2015). Recent and noteworthy review of familial pancreatic cancer genetics, risk assessements and strategies for management.
Tomasetti, C. & Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015).
Sangiorgi, E. & Capecchi, M. R. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc. Natl Acad. Sci. USA 106, 7101–7106 (2009).
Furuyama, K. et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet. 43, 34–41 (2011).
Wu, S., Powers, S., Zhu, W. & Hannun, Y. A. Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47 (2016).
Roberts, N. J. et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 6, 166–175 (2016). Most comprehensive study to date of the germline alterations in a large cohort of patients with familial pancreatic cancer.
Bunting, S. F. & Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 13, 0443–454 (2013).
Willis, N. A. et al. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559 (2014).
Kinzler, K. W. & Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 386, 761–763 (1997). Defined how mutations in gatekeeper genes directly affect tumour growth whereas mutations in caretaker genes create instability that indirectly affects tumour growth.
Stolzenberg-Solomon, R. Z. & Amundadottir, L. T. Epidemiology and inherited predisposition for sporadic pancreatic adenocarcinoma. Hematol. Oncol. Clin. North Am. 29, 619–640 (2015).
Weiss, F. U. Pancreatic cancer risk in hereditary pancreatitis. Front. Physiol. 5, 70 (2014).
Lee, K. E. & Bar-Sagi, D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 18, 448–458 (2010). Provided a mechanism by which oncogenic KRAS bypasses senescence to initiate pancreatic cancer.
Blackford, A. et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 69, 3681–3688 (2009).
Gukovsky, I., Li, N., Todoric, J., Gukovskaya, A. & Karin, M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 144, 1199–1209 (2013).
Pannala, R., Basu, A., Petersen, G. M. & Chari, S. T. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 10, 288–295 (2009).
Micucci, C., Valli, D., Matacchione, G. & Catalano, A. Current perspectives between metabolic syndrome and cancer. Oncotarget http://dx.doi.org/10.18632/oncotarget.8341 (2016).
Petersen, G. M. et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat. Genet. 42, 224–228 (2010).
Wolpin, B. M. et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat. Genet. 46, 994–1000 (2014).
Childs, E. J. et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat. Genet. 47, 1911–916 (2015).
Kimura, W. How many millimeters do atypical epithelia of the pancreas spread intraductally before beginning to infiltrate? Hepatogastroenterology 50, 2218–2224 (2003).
Hruban, R. H., Goggins, M., Parsons, J. & Kern, S. E. Progression model for pancreatic cancer. Clin. Cancer Res. 6, 2969–2972 (2000). Defined the progression model of pancreatic cancer, beginning with precursor lesions that evolve into invasive carcinoma.
Kanda, M. et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 142, 2730–733 (2012).
Wilentz, R. E. et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 58, 4740–4744 (1998).
DiGiuseppe, J. A. et al. Overexpression of p53 protein in adenocarcinoma of the pancreas. Am. J. Clin. Pathol. 101, 4684–688 (1994).
Wilentz, R. E. et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 60, 2002–2006 (2000).
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 2027–40 (2011). Landmark study that defined the genomic features of chromothripsis, a mechanism supporting punctuated evolution in cancer.
Waddell, N. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501 (2015).
Jones, S. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 (2008).
Biankin, A. V. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012).
Witkiewicz, A. K. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 6, 6744 (2015). References 42–45 defined the genomic landscape of pancreatic cancer, the core pathways targeted by somatic alterations and the multiple degrees of structural variation among tumours.
Murphy, S. J. et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 145, 1098–1109 (2013).
Yachida, S. et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long term survivors. Clin. Cancer Res. 18, 6339–6347 (2012).
Pylayeva-Gupta, Y., Grabocka, E. & Bar-Sagi, D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 1761–774 (2011).
Lito, P., Solomon, M., Li, L.-S., Hansen, R. & Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608 (2016).
Patricelli, M. P. et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 6, 316–329 (2016).
Calhoun, E. S. et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am. J. Pathol. 163, 1255–1260 (2003).
Kim, W. Y. & Sharpless, N. E. The regulation of INK4/ARF in cancer and aging. Cell 127, 265–275 (2006).
Quelle, D. E., Zindy, F., Ashmun, R. A. & Sherr, C. J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83, 993–1000 (1995).
Schutte, M. et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 57, 3126–3130 (1997).
Sharpless, N. E. & DePinho, R. A. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 9, 22–30 (1999).
Bertoli, C., Skotheim, J. M. & de Bruin, R. A. M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell. Biol. 14, 518–528 (2013).
Meeker, A. K. et al. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin. Cancer Res. 10, 3317–3326 (2004).
Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53 network. Nature 408, 307–310 (2000).
Hermanova, M. et al. Clinicopathological correlations of cyclooxygenase-2, MDM2, and p53 expressions in surgically resectable pancreatic invasive ductal adenocarcinoma. Pancreas 38, 565–571 (2009).
Roberts, N. J. et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2, 41–46 (2012).
Herbig, U., Jobling, W. A., Chen, B. P. C., Chen, D. J. & Sedivy, J. M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, 53, and p21CIP1, but not p16INK4a. Mol. Cell 14, 501–513 (2004).
Hahn, S. A. et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271, 350–353 (1996).
Shi, Y. & Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003).
Siegel, P. M. & Massagué, J. Cytostatic and apoptotic actions of TGFβ in homeostasis and cancer. Nat. Rev. Cancer 3, 807–821 (2003).
Whittle, M. C. et al. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell 161, 1345–1360 (2015).
Collisson, E. A. et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 17, 500–503 (2011).
Moffitt, R. A. et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178 (2015).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). References 66–68 described phenotypic subtypes of pancreatic cancer based on expression profiling.
Crane, C. H. & Iacobuzio-Donahue, C. A. Keys to personalized care in pancreatic oncology. J. Clin. Oncol. 30, 4049–4050 (2012).
Saha, D., Datta, P. K. & Beauchamp, R. D. Oncogenic ras represses transforming growth factor-β/Smad signaling by degrading tumor suppressor Smad4. J. Biol. Chem. 276, 29531–29537 (2001).
Kretzschmar, M., Doody, J., Timokhina, I. & Massagué, J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev. 13, 804–816 (1999).
Cordenonsi, M. et al. Integration of TGF-β and Ras/MAPK signaling through p53 phosphorylation. Science 315, 843 (2007).
Iglesias, M., Frontelo, P., Gamallo, C. & Quintanilla, M. Blockade of Smad4 in transformed keratinocytes containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene 19, 4134–4145 (2000).
Cordenonsi, M. et al. Links between tumor suppressors: p53 is required for TGF-β gene responses by cooperating with Smads. Cell 113, 301–314 (2003).
Adorno, M. et al. A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137, 87–98 (2009).
Rubinson, D. A. & Wolpin, B. M. Therapeutic approaches for metastatic pancreatic adenocarcinoma. Hematol. Oncol. Clin. North Am. 29, 761–776 (2015).
Richardson, D. M. & Pyšek, P. Naturalization of introduced plants: ecological drivers of biogeographical patterns. New Phytol. 196, 383–396 (2012).
Suarez, A. V. & Tsutsui, N. D. The evolutionary consequences of biological invasions. Mol. Ecol. 17, 351–360 (2008).
Rhim, A. D. et al. EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012).
Stromnes, I. M., DelGiorno, K. E., Greenberg, P. D. & Hingorani, S. R. Stromal reengineering to treat pancreas cancer. Carcinogenesis 35, 1451–1460 (2014).
Neesse, A., Algül, H., Tuveson, D. A. & Gress, T. M. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 64, 1476–1484 (2015).
Cohen, R. et al. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 6, 16832–16847 (2015).
Sousa, C. M. & Kimmelman, A. C. The complex landscape of pancreatic cancer metabolism. Carcinogenesis 35, 1441–1450 (2014).
Vonderheide, R. H. & Bayne, L. J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 25, 200–205 (2013).
Ceyhan, G. O. & Friess, H. Pancreatic disease in 2014: pancreatic fibrosis and standard diagnostics. Nat. Rev. Gastroenterol. Hepatol 12, 68–70 (2015).
Gurtner, G. C., Werner, S., Barrandon, Y. & Longaker, M. T. Wound repair and regeneration. Nature 453, 314–321 (2008).
Dvorak, H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
Masamune, A. & Shimosegawa, T. Pancreatic stellate cells: a dynamic player of the intercellular communication in pancreatic cancer. Clin. Res. Hepatol Gastroenterol. 39 (Suppl. 1), S98–S103 (2015).
Bailey, J. M. et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 14, 5995–6004 (2008).
Bachem, M. G. et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921 (2005).
Taeger, J. et al. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol. Cancer Ther. 10, 2157–2167 (2011).
Sherman, M. H. et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93 (2014).
Olive, K. P. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009). Demonstrated that poor vascularization and perfusion may contribute to inefficient therapy delivery in pancreatic tumours, leading to primary resistance.
Provenzano, P. P. et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429 (2012).
Jacobetz, M. A. et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120 (2013).
Rhim, A. D. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747 (2014).
Özdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
White, E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27, 2065–2071 (2013).
Flajnik, M. F. & Kasahara, M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 11, 47–59 (2010).
Clark, C. E. et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 67, 9518–9527 (2007). Characterized the immune response during tumour evolution using a mouse model of pancreatic cancer.
Feig, C. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl Acad. Sci. USA 110, 20212–20217 (2013).
Beatty, G. L. et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616 (2011).
Foley, K., Kim, V., Jaffee, E. & Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. http://dx.doi.org/10.1016/j.canlet.2015.12.020 (2015).
Fidler, I. J. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2′-deoxyuridine. J. Natl Cancer Inst. 45, 773–782 (1970).
Maddipati, R. & Stanger, B. Z. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov. 5, 1086–1097 (2015).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014).
Maire, F. et al. Differential diagnosis between chronic pancreatitis and pancreatic cancer: value of the detection of KRAS2 mutations in circulating DNA. Br. J. Cancer 87, 551–554 (2002).
Schubbert, S., Shannon, K. & Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308 (2007).
Pylayeva-Gupta, Y., Lee, K. E., Hajdu, C. H., Miller, G. & Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847 (2012).
Zhang, Y. et al. CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol. Res. 2, 423–435 (2014).
McAllister, F. et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25, 621–637 (2014).
Morton, J. P. et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl Acad. Sci. USA 107, 246–251 (2010).
Hitchins, M. P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 15, 625–634 (2015).
Cui, X.-J. et al. High-dose aspirin consumption contributes to decreased risk for pancreatic cancer in a systematic review and meta-analysis. Pancreas 43, 135–140 (2014).
Shain, A. H. et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl Acad. Sci. USA 109, E252–E259 (2012).
Masliah-Planchon, J., Bièche, I., Guinebretière, J.-M., Bourdeaut, F. & Delattre, O. SWI/SNF chromatin remodeling and human malignancies. Annu. Rev. Pathol. 10, 145–171 (2015).
Rao, R. C. & Dou, Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 15, 334–346 (2015).
Bitler, B. G. et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 21, 231–238 (2015).
Helming, K. C. et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 20, 251–254 (2014).
Kaufman, B. et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 33, 244–250 (2015).
Robertson-Tessi, M., Gillies, R. J., Gatenby, R. A. & Anderson, A. R. A. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res. 75, 1567–1579 (2015).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Snyder, A., Wolchok, J. D. & Chan, T. A. Genetic basis for clinical response to CTLA-4 blockade. N. Engl. J. Med. 372, 783 (2015).
Castro-Giner, F., Ratcliffe, P. & Tomlinson, I. The mini-driver model of polygenic cancer evolution. Nat. Rev. Cancer 15, 680–685 (2015).
Wang, L. et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 22, 208–219 (2012).
Haeno, H. et al. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 148, 362–375 (2012).
Gillies, R. J., Verduzco, D. & Gatenby, R. A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 12, 487–493 (2012).
Niklas, K. J. The evolutionary-developmental origins of multicellularity. Am. J. Bot. 101, 6–25 (2014).
Niklas, K. J. & Newman, S. A. The origins of multicellular organisms. Evol. Dev. 15, 41–52 (2013).
Rokas, A. The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu. Rev. Genet. 42, 235–251 (2008).
Domazet-Lošo, T. et al. Naturally occurring tumours in the basal metazoan Hydra. Nat. Commun. 5, 4222 (2014).
Gateff, E. Malignant neoplasms of genetic origin in Drosophila melanogaster. Science 200, 1448–1459 (1978).
Natarajan, L. C., Melott, A. L., Rothschild, B. M. & Martin, L. D. Bone cancer rates in dinosaurs compared with modern vertebrates. Trans. Kans. Acad. Sci. 110, 155–158 (2007).
Tian, X. et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349 (2013).
Abegglen, L. M. et al. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA 314, 1850–1860 (2015).
Vasseur, E. & Quintana-Murci, L. The impact of natural selection on health and disease: uses of the population genetics approach in humans. Evol. Appl. 6, 596–607 (2013).
Matthaei, H., Schulick, R. D., Hruban, R. H. & Maitra, A. Cystic precursors to invasive pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol 8, 141–150 (2011).
Chantrill, L. A. et al. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin. Cancer Res. 21, 2029–2037 (2015).
Hoadley, K. A. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014).
Rech, A. J. et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl Med. 4, 134ra62 (2012).
Soares, K. C. et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 38, 1–11 (2015).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013).
Acknowledgements
This work was supported by CA179991 (C.A.I.-D.), Melanoma Research Alliance #305021 (C.A.I.-D.), F31 CA180682 (A.M.-M.) and T32 CA160001 (A.M.-M.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Driver gene
-
A gene that confers a selective growth or survival advantage when somatically mutated.
- Tree of Life
-
Standard illustration of branching evolution encompassing the history of life on Earth in which branch tips represent extinct or extant species and nodes depict common ancestors.
- Familial pancreatic cancer
-
Pancreatic cancer diagnosis in two or more first-degree relatives.
- Caretaker genes
-
Genes that when mutated result in a loss of genomic stability and fidelity.
- Gatekeeper gene
-
A gene that when mutated results in a loss of growth control.
- Mitotic figures
-
Visible, organized chromosomes in a cell, used as evidence of active mitosis.
- Pseudopapillary
-
Having round outgrowths of tumour cells into the lumen of an epithelial cell-lined duct.
- Fold-back inversions
-
Chromosomal mutations involving a duplication of a genetic sequence followed by an inversion of the copy, resulting in a head-to-head rearrangement.
- Receptor SMADs
-
Transcription factors that are activated by extracellular ligands to promote transforming growth factor-β (TGFβ) signalling and gene expression.
- α-Smooth muscle actin (αSMA)+ myofibroblasts
-
Collagen-producing cells of mesodermal origin that express αSMA when activated.
- Pancreatic stellate cells
-
(PSCs). Resident cells of the pancreas that generate fibrous extracellular matrix.
- Allopatric evolution
-
A type of evolutionary divergence that occurs when a side population is separated from the ancestral population by a physical barrier.
- CD4+ forkhead box P3 (FOXP3)+ regulatory T cells
-
(Treg cells) Immune cells that maintain self-tolerance and suppress immunological response.
- M2 tumour-associated macrophages
-
(TAMs). Immune cells found in pancreatic tumours that promote inflammation.
- Myeloid-derived suppressor cells
-
(MDSCs). Immune cells of myeloid origin that regulate the immune response.
- Fibroblast activation protein (FAP)+ fibroblasts
-
Stromal cells that commonly react with tumour cells.
- Constitutional epimutation
-
A stably inherited epigenetic alteration that leads to changes in gene expression.
- SWI/SNF complex
-
Evolutionarily ancient group of proteins that remodel chromatin by altering the positions of nucleosome binding.
- Immunoediting
-
Immunological selection imposed on tumour cells that may result in the emergence of immune-resistant tumour cells.
- Mini-driver model of polygenic cancer evolution
-
Model of cancer progression in which mutations with subtle effects may collectively confer a survival growth advantage.
- Haploinsufficiency
-
Occurs when one functional copy of a gene is present but abnormal expression or phenotype still occurs.
Rights and permissions
About this article

Cite this article
Makohon-Moore, A., Iacobuzio-Donahue, C. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer 16, 553–565 (2016). https://doi.org/10.1038/nrc.2016.66
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrc.2016.66
This article is cited by
-
A novel cuproptosis-related gene model predicts outcomes and treatment responses in pancreatic adenocarcinoma
BMC Cancer (2023)
-
SETD8 inhibits ferroptosis in pancreatic cancer by inhibiting the expression of RRAD
Cancer Cell International (2023)
-
The genetic landscape of pancreatic head ductal adenocarcinoma in China and prognosis stratification
BMC Cancer (2022)
-
Integration of metabolites from meta-analysis with transcriptome reveals enhanced SPHK1 in PDAC with a background of pancreatitis
BMC Cancer (2022)
-
Fbxo45 facilitates pancreatic carcinoma progression by targeting USP49 for ubiquitination and degradation
Cell Death & Disease (2022)
