
Abstract
A central question in evolutionary biology is whether sponges or ctenophores (comb jellies) are the sister group to all other animals. These alternative phylogenetic hypotheses imply different scenarios for the evolution of complex neural systems and other animal-specific traits1,2,3,4,5,6. Conventional phylogenetic approaches based on morphological characters and increasingly extensive gene sequence collections have not been able to definitively answer this question7,8,9,10,11. Here we develop chromosome-scale gene linkage, also known as synteny, as a phylogenetic character for resolving this question12. We report new chromosome-scale genomes for a ctenophore and two marine sponges, and for three unicellular relatives of animals (a choanoflagellate, a filasterean amoeba and an ichthyosporean) that serve as outgroups for phylogenetic analysis. We find ancient syntenies that are conserved between animals and their close unicellular relatives. Ctenophores and unicellular eukaryotes share ancestral metazoan patterns, whereas sponges, bilaterians, and cnidarians share derived chromosomal rearrangements. Conserved syntenic characters unite sponges with bilaterians, cnidarians, and placozoans in a monophyletic clade to the exclusion of ctenophores, placing ctenophores as the sister group to all other animals. The patterns of synteny shared by sponges, bilaterians, and cnidarians are the result of rare and irreversible chromosome fusion-and-mixing events that provide robust and unambiguous phylogenetic support for the ctenophore-sister hypothesis. These findings provide a new framework for resolving deep, recalcitrant phylogenetic problems and have implications for our understanding of animal evolution.
Similar content being viewed by others

Main
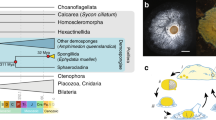
Five major lineages arose early in animal evolution and survive to the present day: sponges (poriferans), ctenophores (comb jellies), placozoans (microscopic flat animals), cnidarians (such as anemones, jellyfishes and hydra) and bilaterians (such as chordates, molluscs, arthropods and diverse worms)1,8,10,13,14. Although morphological and phylogenomic studies consistently unite bilaterians, cnidarians, and placozoans into a monophyletic clade (Parahoxozoa) that excludes sponges and ctenophores8,10,14 the relationship between sponges, ctenophores and Parahoxozoa remains controversial. There are two competing scenarios—the sponge-sister hypothesis7,8 and the ctenophore-sister hypothesis9,10—reflecting which lineage diverged first among animals (Fig. 1a).

a, Two alternative metazoan phylogenetic hypotheses, with either ctenophores (left) or sponges (right) as sister to all other animals. b,c, Specimens of species of which the genomes are reported here. Scale bars, 1 cm. b, The lobate ctenophore B. microptera from the Monterey Bay, California. c, Undescribed cladorhizid demosponge collected offshore of Big Sur, California at a depth of 3,975 m. d, Ribbon diagram showing conserved syntenies among animals (α ≤ 0.05, permutation test one-sided false-discovery rate), including (from top to bottom) two ctenophores (B. microptera (BMI) and H. californensis); the jellyfish R. esculentum; the bilaterian amphioxus B. floridae; and two demosponges (E. muelleri and the cladorhizid demosponge). Each horizontal black bar represents a chromosome. The vertical lines between species represent orthologous genes, coloured according to the BCnS synteny groups12. Only groups of genes that have significantly conserved chromosome-scale linkage (synteny) between metazoan species are shown. There is extensive 1:1 conserved chromosomal synteny between the two ctenophores, consistent with the conserved ctenophore karyotype. Orthologous gene pairs in the two ctenophores that do not participate in conserved syntenies with BCnS are shown in grey. Photography credits: Shannon Johnson © 2019 MBARI (b), © 2021 MBARI (c).
As sponges and ctenophores are such disparate animals13, the nature of the first diverging animal lineage has implications for the evolution of fundamental animal characteristics. Adult sponges are generally sessile filter-feeding organisms with body plans organized into reticulated water-filtration channels, structures built out of silica or calcium carbonate, and specialized cell types and tissues used for feeding, reproduction and self-defence, but they lack neuronal and muscle cells15. By contrast, ctenophores are gelatinous marine predators that move using eight longitudinal ‘comb rows’ of ciliary bundles16,17; they are superficially similar but unrelated to cnidarian medusae13,18 and possess multiple nerve nets19. Thus, whereas the sponge-sister scenario suggests a single origin of neurons on the ctenophore–parahoxozoan stem, the ctenophore-sister scenario implies either that either ancestral metazoan neurons were lost in the sponge lineage, or that there was convergent evolution of neurons in the ctenophore and parahoxozoan lineages3,6. Similar considerations apply to other metazoan cell types18, gene regulatory networks, animal development13,18 and other uniquely metazoan features.
Despite its importance for understanding animal evolution, the relative branching order of sponges, ctenophores and other animals has proven to be difficult to resolve2. The fossil record is largely silent on this issue as verified Precambrian sponge fossils are extremely rare20 and putative fossils of the soft-bodied ctenophores are difficult to interpret21. Morphological characters of living groups (for example, choanocytes of sponges) are not sufficient to resolve the question because true homology is difficult to assign, and such characters are easily lost or can arise convergently13,22. The ctenophore-sister hypothesis is supported by a pair of gene duplications shared by sponges, bilaterians, placozoans and cnidarians but not ctenophores23. Although sophisticated methods for sequence-based phylogenomics have been developed and applied to increasingly large molecular datasets, there is still considerable debate about the relative position of sponges and ctenophores as results are sensitive to how sequence evolution is modelled11, which taxa or sites are included24,25, and the effects of long-branch artifacts and nucleotide compositional variation26. New approaches are needed.
We reasoned that patterns of synteny, classically defined as chromosomal gene linkage without regard to gene order27, could provide a powerful tool for resolving the ctenophore-sister versus sponge-sister debate. Chromosomal patterns of gene linkage evolve slowly in many lineages12,28,29,30, probably because it is improbable for interchromosomal translocations to be fixed in populations with large effective population sizes28,31,32. Notably, some changes in synteny are effectively irreversible. For example, when two distinct ancestral synteny groups are combined onto a single chromosome by translocation, and subsequent intrachromosomal rearrangements mix these two groups of genes, it is very unlikely that the ancestral separated pattern will be restored by further rearrangement and fission, in the same sense that spontaneous reduction in entropy is improbable12. Such rare and irreversible changes are particularly useful for resolving challenging phylogenetic questions as they give rise to shared derived features that unambiguously unite all descendant lineages33,34,35. Deeply conserved syntenies observed between animals and their closest unicellular relatives12 suggest that outgroup comparisons could be used to infer ancestral metazoan states and polarize changes within animals to address the sponge-sister versus ctenophore-sister debate. Yet, chromosome-scale genome sequences of the unicellular or colonial eukaryotic outgroups closest to animals (choanoflagellates, filastereans and ichthyosporeans) have not been reported.
Here we show that conserved syntenies between animals and their closest unicellular relatives support ctenophores as the sister group to all other animals. Specifically, we find seven sets of genes for which (1) ctenophores share ancestral metazoan gene linkages with one or more unicellular eukaryotes; and (2) bilaterians, cnidarians, placozoans and sponges are united (to the exclusion of ctenophores) by shared derived patterns of synteny that arose by ancient interchromosomal translocations. In four of these cases, irreversible mixing after chromosome fusion evidently occurred on the bilaterian–cnidarian–sponge (BCnS) stem lineage, providing unambiguous support for the ctenophore-sister scenario. The alternative sponge-sister hypothesis is not supported by any synteny-based characters, and would require reversal of four sets of fusion-with-mixing events and/or extensive convergent fusion in both sponges and on the bilaterian/cnidarian stem to account for the observed patterns of synteny. To enable these analyses, we generated chromosome-scale genome sequences for three animal species (two sponges and a ctenophore), and three non-animal species (a filasterean, ichthyosporean and choanoflagellate) to serve as outgroups. Our analyses further reveal ancient syntenies conserved between animals and their closest unicellular relatives (animal plesiomorphies) as well as metazoan syntenies shared by all animals but not present in unicellular organisms (animal synapomorphies). These findings establish a phylogenetic framework for understanding the early evolution of metazoan genomes and characters.
To examine conserved syntenies across animals, we traced the chromosomal distribution of orthologous genes among diverse metazoan lineages using previously and newly sequenced genomes (Fig. 1bc, Methods, Supplementary Information 1–3 and Supplementary Data 1). Figure 1d highlights conserved metazoan synteny groups, that is, groups of genes of which orthologues are linked on the same chromosome across multiple lineages, regardless of gene order. Syntenic groups shown in Fig. 1d are statistically significant (Methods). In Fig. 1d, lines connecting orthologous genes are coloured according to the previously identified BCnS ancestral linkage groups (ALGs)12,28,29,30. For example, the group on the far left represents the BCnS ALG_M (comprising genes found on jellyfish Rhopilema esculentum chromosome 2 (RES2), amphioxus Branchiostoma floridae chromosome 8 (BFL8), and sponge chromosomes CLA16 (of a cladorhizid demosponge) and EMU16 (of Ephydatia muelleri)). Note that, by our definition, two different conserved synteny groups can coexist on the same chromosome in some species. For example, amphioxus chromosome BFL5 is seen to be a combination of BCnS ALGs Ea and Eb, which are found on distinct chromosomes in other species.
Our results extend previous findings12 of BCnS ALGs by incorporating a new chromosome-scale genome sequence of a recently discovered bioluminescent deep-sea cladorhizid demosponge36 (Fig. 1c, Supplementary Information 2 and Extended Data Fig. 1c–i) complementing the spongillid demosponge E. muelleri37. Although the cladorhizid and spongillid lineages diverged approximately 450 million years ago38, chromosomes of the two demosponges correspond simply with each other (Fig. 1d and Extended Data Fig. 2f–h) and with bilaterian and cnidarian chromosomes (Fig. 1d and Extended Data Fig. 2i–k), consistent with the previously described genome tectonic schema12. Further comparisons with other recently released chromosome-scale demosponge genome sequences39 confirm the high degree of conserved synteny in this group, but show that one of the rearrangements that we found in the cladorhizid genome is the result of a fission in that lineage (ALG_H; Extended Data Fig. 2f–k). We also sequenced the genome of a previously undescribed hexactinellid (glass) sponge (Extended Data Fig. 1j–n and Supplementary Information 2), but found it to be considerably rearranged. Despite many lineage-specific genomic changes in glass sponges, relicts of 10 out of 29 BCnS ALGs are detectable (Extended Data Fig. 3). Owing to the high degree of rearrangement, we do not consider hexactinellid genomes further.
However, in contrast to demosponges, genomic comparisons between the cydippid ctenophore Hormiphora californensis40 and other metazoans reveal patterns of both conserved and altered synteny (Fig. 1d and Extended Data Fig. 2b–e). For example, whereas the BCnS group ALG_Ea is localized to a single ctenophore chromosome (H. californensis chromosome 8 (HCA8)), the BCnS synteny group ALG_A1a (comprising genes found on amphioxus chromosome BFL1, jellyfish chromosome RES2, and sponge chromosomes EMU1 and CLA15) is partitioned across two ctenophore chromosomes (HCA12 and HCA7). To test whether the observed patterns of ctenophore synteny are unique to the H. californensis lineage or common across ctenophores, we assembled and analysed the genome of the recently redescribed41 lobate comb jelly Bolinopsis microptera (Fig 1b; the assembly is reported in the Methods, Supplementary Information 1, Extended Data Fig. 1a,b and Supplementary Table 1.1–1.4). Despite the 160–260-million-year divergence between lobate and cydippid ctenophores10 their n = 13 chromosomes show one-to-one correspondence (without gene order conservation) (Fig. 1d and Extended Data Fig. 2a). This finding implies that a common n = 13 karyotype is ancestral for the Hormiphora–Bolinopsis crown group, and that cross-metazoan patterns of synteny shown in Fig. 1d are general.
Interpreting the differences in synteny between ctenophores and other animals depends on the ancestral metazoan state (Fig. 2c–g, Extended Data Fig. 4 and Supplementary Information 4). If BCnS syntenies are ancestral to all metazoans, then the partitioned syntenies observed in ctenophores would have arisen by rearrangements that split the ancestral chromosomes in the ctenophore lineage (syntenic autapomorphies; Fig. 2e) and would therefore be uninformative for discriminating between the ctenophore-sister and sponge-sister hypotheses. Alternatively, if the patterns of synteny found in ctenophores are ancestral to animals, the derived syntenies shared by BCnS to the exclusion of ctenophores could have arisen by fusion on the BCnS stem lineage, which would represent syntenic synapomorphies (Fig. 2f,g). In this case, ctenophores would be excluded from the BCnS clade and established as the sister clade of all other extant metazoans. Note that the extensive conservation of synteny between sponges, bilaterians, and cnidarians12 confirmed here makes it improbable that ctenophores could share syntenies with cnidarians and bilaterians to the exclusion of sponges and, indeed, we did not find any such cases in analyses described below.

a,b, Conserved linkages between the chromosomes of animals and two non-animal outgroups (α ≤ 0.05, permutation test one-sided false-discovery rate). a, The filasterean amoeba C. owczarzaki. b, The choanoflagellate S. rosetta. Each synteny group (conserved between metazoans and an outgroup) was assigned a distinct colour (different from Fig. 1). c–g, Schematics showing phylogenetic information (ancestor to OABC (c); ancestral fusion or outgroup fission (d); derived fusion (e); fusion and lineage-specific mixing (f); and irreversible fusion with mixing (g)) conveyed by patterns of conserved synteny based on a quartet analysis. Node O designates the outgroup, and nodes A, B and C are ingroups of which the phylogenetic branching is to be determined. The thin red and blue lines in d–g represent genes of distinct synteny groups on different chromosomes in at least one species. Changes in syntenic characters are indicated schematically on parsimonious phylogenies on the left of each diagram. d,e, Single-species differences are phylogenetically uninformative. f,g, Shared chromosomal distributions between outgroup O and one of the ingroups (labelled taxon A) imply that the other two ingroups (taxa B and C) are related by fusion of ancestral synteny groups. f and g differ in whether all fusions have subsequently mixed. Fusion with mixing (g) is the strongest phylogenetic character because it represents an irreversible change, as discussed in the main text. h,i, Subsets of the synteny groups shown in a and b (Capsaspora (h) and Salpingoeca (i)) that match the phylogenetically informative patterns indicated in f and g. In all such cases, ctenophore syntenies match the outgroup and sponges share fusions with bilaterians and cnidarians. Note that groups A1a and G are found in both outgroups. We did not observe any cases in which sponge syntenies match the outgroup to the exclusion of ctenophores, bilaterians and cnidarians.
To provide outgroups for inferring ancestral metazoan syntenies, we assembled chromosome-scale sequences of representatives of three unicellular lineages closest to animals (collectively, outgroups): the choanoflagellate Salpingoeca rosetta (chromosome number, n = 36), the filasterean amoeba Capsaspora owczarzaki (chromosome number, n = 16) and the ichthyosporean Creolimax fragrantissima (chromosome number, n = 26). Chromosome-scale sequences and karyotypes were obtained by integrating previously reported subchromosomal draft sequences42,43,44 with new chromatin conformation data (Methods, Supplementary Information 3 and Extended Data Fig. 5).
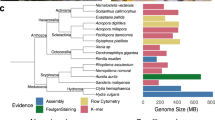
Chromosomal comparisons across animal and non-animal outgroup genomes revealed conserved ancestral metazoan synteny groups ranging in size from 5 to 29 genes, totalling 291 genes (out of 2,474 outgroup-metazoan orthology groups; Methods, Figs. 2 and 3, Extended Data Tables 1 and 2, Extended Data Figs. 5–8, Supplementary Information 5–9 and Supplementary Data 2). This finding extends previous observations based on subchromosomal assemblies of non-animal species12. Each such ancestral metazoan synteny group is a collection of genes of which the orthologues are consistently linked on single chromosomes in diverse metazoans and at least one outgroup (Fig. 2h–i and Extended Data Table 2). In contrast to the readily detected conserved syntenies among sponges, cnidarians and bilaterians, conserved syntenies involving ctenophores and non-animal outgroups are not visually evident in pairwise comparisons with other animals (Extended Data Figs. 2 and 6) but are statistically supported in multispecies comparisons (Methods and Supplementary Information 4 and 11). On the basis of permutation tests, the false-discovery rate of a conserved group of five linked genes in a four-species comparison is α ≤ 0.0003, and groups of eight or more linked genes never occurred in ten million permutations (Supplementary Information 8 and Extended Data Table 2). To maximize coverage of lineages relevant for the branching order of sponges and ctenophores, we considered orthologous genes across quartets of the form {outgroup, sponge, ctenophore, cnidarian/bilaterian}, which does not presuppose either the ctenophore-sister or sponge-sister hypothesis (Fig. 3 and Supplementary Information 4, 8 and 9). The extensive conservation of synteny across BCnS and within ctenophores makes our analysis insensitive to which genomes are used to represent these major metazoan clades (Supplementary Information 8). Here we used the scallop Pecten maximus45, the fire jellyfish R. esculentum46, the freshwater sponge E. muelleri37 and the ctenophore H. californensis to represent the bilaterian, cnidarian, sponge and ctenophore genomes (Methods), although our findings do not depend on these choices (Figs. 2 and 3). We used two different methods for identifying orthologues—a simple mutual-best-hits method and an alternative orthologue-clustering approach (OrthoFinder47; Supplementary Information 10 and 11), and obtained comparable results using both approaches (Extended Data Fig. 9).

a, The rows represent the species considered in our analyses. The top three rows are non-metazoan outgroups. Columns show pairs of phylogenetically informative and significantly large metazoan syntenies (α ≤ 0.05, permutation test one-sided false-discovery rate), labelled according to their BCnS names (A1a, C1 and so on) with the suffix _x or _y denoting subgroups shared across metazoans. The number of genes participating in each metazoan synteny group is indicated in red and blue rectangles at the top of each column. Only genes with defined orthologues in outgroups are shown here. Extended Data Fig. 8 shows a larger set of genes requiring only metazoan orthologues. Inset: the convention for representing gene distributions on chromosomes (top left). The grey rectangles represent chromosomes (or large scaffolds in the placozoan Trichoplax). The chromosome number or scaffold name is located above or to the left of the grey rectangle. Red and blue vertical hashes represent the relative position of genes participating in phylogenetically informative pairs of metazoan synteny groups. b, The most parsimonious phylogeny according to the logic of Fig. 2c–g (Extended Data Fig. 4 and Supplementary Information 4), the results of ref. 12 and the accepted monophyly of demosponges.
Although choanoflagellates are considered to be the closest living relatives of animals48,49, we found that the more distantly related filasterean Capsaspora shares 29 conserved synteny groups with metazoans, compared to 20 between the choanoflagellate Salpingoeca and metazoans (Extended Data Figs. 6 and 7), perhaps indicating more rapid interchromosomal rearrangement in the Salpingoeca lineage. The even more distantly related ichthyosporean Creolimax still retains eight conserved synteny groups with metazoans. Although we considered each outgroup-plus-metazoan comparison separately, we found widespread overlap between the ancient synteny groups defined independently by comparison with Capsaspora and Salpingoeca. In total, our analysis defined 31 ancestral metazoan synteny groups that are traceable to the last common ancestor of Metazoa and shared by one or both of Capsaspora and Salpingoeca (Extended Data Table 1). The extensive conservation of synteny within BCnS implies that the ancestral metazoan synteny groups correspond to subsets of the BCnS groups, and we name them using the BCnS notation with the suffixes _x and _y. If we relax the condition that an outgroup gene must be present, more metazoan genes can be added to these ancestral metazoan syntenic units (Extended Data Fig. 8).
Conservation of synteny between animals and their unicellular relatives may at first seem surprising, as these lineages diverged more than 800 million years ago50. Within animals, it has been estimated that ongoing small-scale translocations between chromosomes typically transfer 1% of genes to a different chromosome every ~40 million years12. The limited residual conservation of synteny between animals and close unicellular relatives suggests that small-scale translocations have occurred at similarly low rates along both choanoflagellate and filasterean lineages. The more extensive conservation observed between animals and Capsaspora versus Salpingoeca may be due to variations in this rate or differences in other chromosomal rearrangements over deep time. The Capsaspora karyotype is predominantly metacentric and, notably, we find that 11 of the 29 ancient synteny groups found in Capsaspora are concentrated on single chromosome arms, rather than dispersed across whole Capsaspora chromosomes, based on estimates of centromere position using chromatin conformation contacts. This raises the possibility that Capsaspora chromosome arms preserve ancient filozoan units and suggests further attention to the chromosome biology of non-metazoan relatives. We found no significant functional associations of anciently linked groups of genes (Supplementary Information 12 and Supplementary Data 3), consistent with a general slow rate of synteny loss due to the infertility of translocation heterozygotes28,31, which allows only small-scale interchromosomal translocation32.
With conserved ancestral metazoan syntenies in hand, we tested the ctenophore-sister versus sponge-sister hypotheses by identifying shared, derived syntenic characters using standard phylogenetic methods. As noted above, two or more metazoan synteny groups can co-occur on the same chromosome in one or more genomes, corresponding to ancient fusions (that is, translocations51) (Fig. 2h,i). As only shared derived characters are phylogenetically informative, changes that are unique to a single lineage can be disregarded (Fig. 2e and Extended Data Fig. 4b,d,e,g). There are two different types of chromosomal fusions between two ancestrally linked groups of genes: without mixing (Fig. 2f) or with subsequent intermixing (Fig. 2g and Supplementary Data 4 and 5). Fusion-without-mixing is potentially reversible, as observed in Robertsonian fusions and fissions involving whole chromosome arms51. However, in the fusion-with-mixing case, reversion is extremely unlikely, comparable to the spontaneous reduction of entropy after mixing of two fluids12.
We encoded the state of each potential fusion into a phylogenetic character matrix as 0 (no fusion, that is, ALGs found on separate chromosomes), 1 (fused but unmixed) or 2 (fused and mixed). The mixed/unmixed status of a fusion was determined on the basis of the likelihood of the observed gene arrangement under a model of random rearrangement (that is, entropy of mixing of the two fused groups) (Methods, Supplementary Information 13 and Supplementary Data 4 and 5). The same fusion character states were obtained using orthology defined by mutual-best-hits or OrthoFinder. We then applied the machinery of Bayesian phylogenetics52 to this character matrix, using asymmetric transition probabilities to reflect the highly improbable unmixing transition (Methods and Supplementary Data 6).
Bayesian phylogenetic analyses of the fusion character matrix strongly support the ctenophore-sister topology (Fig. 4 and Supplementary Information 14). The same conclusion is clear from direct examination of the fusions identified in our data. Specifically, there are seven derived fusions shared by bilaterians, cnidarians and sponges to the exclusion of ctenophores (Extended Data Table 1). Of these seven derived fusions, four are accompanied by mixing of genes from two different ancestral chromosomes—a process that is essentially irreversible (Figs. 2e and 4b–d); the other three are mixed only in bilaterians and cnidarians (Fig. 3).

a, Bayesian phylogenetic analysis of conserved syntenies supports monophyly of the group comprising demosponges, Cnidaria and Bilateria, to the exclusion of Ctenophora, with high posterior probability (red arrow: 1.0, 100,000 generations with 25% burn-in). Bayesian analysis was run on both constrained (per-phylum constrained tree shown) and unconstrained tree topologies (Supplementary Information 14 and Supplementary Data 6). This panel corresponds to Supplementary Fig. 14.1c. b, Character transitions involving ALGs C1, F, L and N in Fig. 3 are most parsimoniously interpreted as fusion with mixing on the myriazoan stem after divergence from ctenophores, which retain the ancestral metazoan state as inferred from outgroup comparisons. c,d, To interpret the observed patterns under the alternative sponge-sister hypothesis would require unlikely convergent chromosomal changes (either convergent fusions (c) or exact unmixing and fissions to the ancestral state (d)) that were not seen in our genomes. e, The number of genes in the genome-shuffling simulations (n = 1 × 108) that support the ctenophore-sister (upper) or sponge-sister (lower) hypothesis. For the ctenophore Hormiphora, the number of fusion-with-mixing events is significantly higher in the observed genomes (vertical red bars) than in the Hormiphora genome-shuffling simulations (vertical grey histogram bars). Significance is shown as the one-sided false-discovery rate, α, of a genome-shuffling permutation test. There were no groups of genes that supported the sponge-sister hypothesis in the real genomes, and none occurred in the genome-shuffling simulations. f, Additional statistical measures also support only the ctenophore (cteno.)-sister hypothesis in genome-shuffling simulations of Hormiphora, Capsaspora (COW), Salpingoeca (SRO), Ephydatia (EMU) and Rhopilema. PI ALG, phylogenetically informative linkage groups. The shape indicates the treatment; the colour indicates the outgroup. The full figure is shown in Extended Data Fig. 10. g, Summary of phylogenetic relationships among animals and close outgroups including syntenic characters. Myriazoa (underlined) is the name proposed for the clade containing extant animals, except Ctenophora. Outgroup topology follows ref. 49.
We reject the alternative sponge-sister hypothesis as it would require either (1) multiple convergent fusions (that is, involving the same groups of genes) in both the sponge and bilaterian-cnidarian lineages (Fig. 4c and Supplementary Information 15) or (2) the precise reversal of multiple fusions-with-mixings in the ctenophore lineage to match the original patterns found in the ancestral metazoan lineage (Fig. 4d). The extreme unlikeliness of recovering the observed syntenic patterns by chance is shown by simulations in which we permuted the configuration of the genes in each of the C. owczarzaki, S. rosetta, H. californensis, E. muelleri and R. esculentum genomes (Supplementary Information 15 and Extended Data Fig. 10). Across one hundred million randomized Hormiphora genomes, we never found syntenic signals comparable to those observed with the actual genome, indicating that syntenic support for ctenophore-sister is unlikely to have arisen by chance (Fig. 4e). We also note the complete absence of syntenic synapomorphies of a hypothetical ctenophore–bilaterian–cnidarian clade that excludes sponges, both in the actual data and in genome-shuffling simulations (Fig. 4f, Extended Data Fig. 10 and Extended Data Tables 1 and 2). This lack of homoplasy allows for a simple interpretation of the results (Fig. 4).
Conclusions
Support for the ctenophore-sister hypothesis
Our findings provide strong support for the ctenophore-sister scenario and reject the sponge-sister hypothesis. Although we encoded syntenic states as a character matrix and analysed it using a Bayesian phylogenetic framework (Fig. 4a and Supplementary Fig. 14.1), the cladistic logic supporting our conclusions is easily appreciated, as emphasized above (Fig. 2, Extended Data Fig. 4 and Supplementary Information 4). Previous phylogenetic analyses of sequence-based characters have not resolved the sponge-sister versus ctenophore-sister hypotheses because the phylogenetic signal is weak and distributed across thousands of individual amino acid positions that are often saturated or subject to confounding evolutionary forces11. By contrast, the synteny-based characters that support ctenophores as sister to other animals in our analysis are clear: sponges, bilaterians, and cnidarians share multiple irreversible changes in synteny to the exclusion of ctenophores (BCnS syntenic synapomorphies) (Figs. 2 and 3). Support for the ctenophore-sister hypothesis is directly testable by future genome sequencing, as it is a strong prediction of our model that all bilaterian, placozoan, cnidarian or sponge genomes should share the four fusion-with-mixing syntenic synapomorphies shown in Figs. 2 and 3 and, to a lesser extent, the three fusion-without-mixing events (pending considerations of sponge monophyly; Supplementary Information 7.2.6). The placement of ctenophores as sister to other animals also rejects the old notion of a Coelenterata clade that would unite ctenophores with cnidarians53.
Myriazoans
The clade containing all sponges, bilaterians, cnidarians and placozoans is diverse, accounting for all living animals other than ctenophores. In recognition of this morphological diversity, we propose that this clade be called Myriazoa, from the Greek myria (extremely great in number) and zoa (animals) (Fig. 4g). While Myriazoa is supported by shared derived chromosomal fusions, there are currently no obvious morphological characters that unite them. The name Benthozoa was proposed for this clade23 on the basis of the inference of a pelagic ancestral metazoan and a derived benthic adult ancestor of the clade sister to ctenophores, but a benthic life history stage may not be a shared derived feature of this clade. In particular, it would be just as parsimonious for the ancestor of Metazoa to have had a benthic stage, and for most ctenophores to have lost it. We therefore prefer to avoid any assumption of the ancestral life history strategy in referring to the clade.
Parahoxozoans, sponges and placozoans
A clade grouping bilaterians, placozoans and cnidarians to the exclusion of sponges and ctenophores54 has been recovered in multiple phylogenetic studies8,10 and is now called Parahoxozoa on the basis of the shared presence of Hox/ParaHox-class genes14. Parahoxozoa is supported in our analysis by the disposition of the ancestral myriazoan linkage groups Ea and G, which are each partitioned across two chromosomes in non-metazoan outgroups and ctenophores. The pre-myriazoan partitions of Ea and G are fused in demosponges and parahoxozoans, but are mixed only in parahoxozoans, providing a candidate parahoxozoan synapomorphy. The most parsimonious interpretation is that fusions forming Ea and G occurred without mixing on the myriazoan stem, a state that is preserved in demosponges, but that mixing occurred on the parahoxozoan stem lineage so that the mixed state is shared by all bilaterians, cnidarians and placozoans. However, a detailed understanding of the history of Ea and G linkages in sponges will require chromosome-scale genome sequences from other sponge classes beyond demosponges and lyssacinosid glass sponges (Supplementary Information 7.2.2, 7.2.5 and 7.2.6). If sponges are monophyletic (as supported by recent phylogenomic studies8,10,55), then the four fusions-with-mixing that are found in demosponges and parahoxozoans must be shared by all sponges. However, if one or more sponge classes branched before the split between the demosponge and parahoxozoan lineages, it is possible that the descendants of the early-branching sponges might not possess one or more of these myriazoan fusions-with-mixing.
Although the subchromosomal assemblies currently available for Trichoplax preclude its full integration into the present analysis, Fig. 3 shows that placozoans share the diagnostic myriazoan fusion-with-mixing characters related to ALG_C1 and the two bilaterian–cnidarian fusions-with-mixing related to ALGs A1a and G. The placozoa-sister-to-other-animals hypothesis56 is rejected by the placement of placozoans within Myriazoa using synteny. It is therefore a strong prediction of our overall approach that chromosome-scale assemblies of placozoans will show that they share the fusions and mixing events that define Myriazoa. Furthermore, we previously showed that cnidarians and placozoans are united as sister lineages to the exclusion of bilaterians and sponges based on the mixing of genes from ALG_Ea and ALG_F found on cnidarian chromosomes and placozoan scaffolds57, consistent with recent gene trees55. These characters do not appear in the present analysis owing to the stringent requirement that syntenies considered here are also preserved in outgroups to Metazoa. If placozoans are nested within Parahoxozoa, homologies between the mouth, gut and nervous systems of cnidarians and bilaterians imply that placozoans are secondarily flattened and have lost an ancestral nervous system, rather than representing the ancestral parahoxozoan state.
Implications for early animal evolution
Finally, we consider implications of the ctenophore-sister hypothesis for early animal evolution1,2. Comparisons among diverse genomes have identified numerous genes that are present in myriazoans but are absent in ctenophores1,5,58. Under the ctenophore-sister scenario, these are most parsimoniously interpreted as arising on the myriazoan stem after the divergence of ctenophores1,5,59, and include genes associated, in bilaterians and cnidarians, with neuronal function1,5,59, development58 and cell adhesion60. However, as gene loss is common throughout animal evolution61, it is also possible that some of these genes were present in the ancestral metazoan but lost in ctenophores. Similarly, some genes are present in ctenophores and parahoxozoans but absent in sponges58, and these must be interpreted as gene losses on the sponge lineage.
Perhaps the most intriguing suite of metazoan characters pertain to neuromuscular systems, which are present in varying complexity in ctenophores, bilaterians, and cnidarians but are absent in sponges3,6,59. In sponge-sister scenarios, these characters are interpreted as being primitively absent, arising after the divergence of sponges on the stem lineage leading to other animals. However, in the ctenophore-sister scenario supported here by deeply conserved syntenies, there are two possible alternatives explaining the evolution of neurons: either complex neural systems arose more than once3,59,62 but were elaborated differently in ctenophores, cnidarians and bilaterians3,4,59, or neuronal cell types were present in the metazoan ancestor but were lost in the sponge lineage4,9,63.
Sponge-sister and ctenophore-sister hypotheses are sometimes erroneously interpreted as suggesting that the most recent common ancestor of animals was sponge-like or ctenophore-like. We must be mindful, however, that the living representatives of sponges, ctenophores, bilaterians and placozoans may be poor surrogates for the earliest members of each stem-lineage, as the crown group of each clade arose hundreds of millions of years after their divergence from each other, let alone from the common metazoan ancestor2. Although living sponges are often defined by the cellular, morphological and developmental characters that they lack relative to other animals, they are complex animals in their own right, successfully adapted to a unique benthic filter-feeding lifestyle13. Consistent with a neuron-bearing metazoan ancestor, sponges possess secretory cell types15 and extensive molecular components associated with presynaptic function that could be derived from a primitive neurosecretory cell. Conversely, the elaborate and divergent nervous systems of living ctenophores, bilaterians, and cnidarians do not represent the stem ancestors of these groups, which would have had very different lifestyles in the Ediacaran. The nervous systems of living ctenophores, cnidarians, and bilaterians each have unique properties6,19,59, and could represent divergent evolution from a simpler neuron-bearing common ancestor. With the ctenophore-sister topology in hand, reconstructing the characters of this metazoan ancestor will require an improved understanding of molecular, cellular and system homologies and specializations across the full range of animal diversity.
Methods
A full description of the methods is provided in the Supplementary Information.
Unicellular outgroup species genome scaffolding
Chromatin conformation capture (Hi-C) libraries were generated64 from frozen cell cultures obtained directly from the American Type Culture Collection. The cultures used were of the species C. owczarzaki (ATCC, 30864), C. fragrantissima (ATCC, PRA-284) and S. rosetta (ATCC, PRA-366). The strains used were the same as those sequenced in the original genome assembly projects for each species42,43,44. The Hi-C libraries were sequenced at a depth of over 500× for each species on the Illumina NovaSeq 6000 system at MedGenome.
Previously published draft genome assemblies42,44 were scaffolded to chromosome-scale using a combination of HiRise (v.Aug2019)65 and SALSA2 (v.2.3)66. The genomes were manually curated using PretextView v.0.2.4 (https://github.com/wtsi-hpag/PretextView), HiGlass v.1.10.010467, Juicebox Assembly Tools (GitHub Commit 46c7ed1105)68, the Juicebox visualization system (v.1.11.08106)69 and artisanal (https://bitbucket.org/bredeson/artisanal/src). For C. owczarzaki, we used the most recent ‘v4’ assembly as input for scaffolding70. The Hi-C data were used as evidence to remove several megabases of the original C. fragrantissima assembly that, after further analysis, appeared to be fungal contaminants. We identified the general location of the centromeres in C. fragrantissima and C. owczarzaki using the Hi-C data as described in Supplementary Information 3.
Sponge and ctenophore genome assembly
Samples of B. microptera41 were collected in Monterey Bay, California (36.63° N, 121.90° W) from surface waters and were reared to an F3 population at the Monterey Bay Aquarium, from which one adult was sequenced. One individual cladorhizid sponge36 was collected off the coast of Big Sur, California (35.49° N, 124° W) from the seafloor at 3,975 m. One hexactinellid ‘tulip’ sponge (HEX) was collected near Southern California (34.57° N, 122.56° W) from the seafloor at 3,852 m. This species of ctenophore, and presumably these species of sponges, are hermaphroditic. Sponge and ctenophore samples were collected under the State of California Department of Fish and Wildlife collecting permits SC-2026 (Bolinopsis) and SC-4029 (sponges).
DNA and RNA were isolated from these species to generate Pacific Biosciences (PacBio) CLR WGS, HiFi WGS libraries or PacBio Iso-Seq libraries at the Brigham Young University DNA Sequencing Center. These libraries were sequenced on the PacBio Sequel II system. Illumina WGS libraries, Chicago libraries and Hi-C libraries were generated at UC Santa Cruz and sequenced at MedGenome on the Illumina HiSeq X system. PacBio WGS library coverage was over 70× for all three species, and Hi-C coverage was over 190× for all three species. Genome sizes were estimated using jellyfish (v.2.2.10)71, then using the resulting spectrum in GenomeScope (v.2)72.
The genome of B. microptera was assembled using wtdbg (v.2.4)73, and the sponge genomes were assembled using hifiasm (v.0.16.1-r375)74. Hi-C reads were mapped using bwa mem (v.0.7.17)75, processed using pairtools (v.0.3.0)76, pairix (v.0.3.7; https://github.com/4dn-dcic/pairix) and Cooler (v.0.8.10)77, and scaffolding was performed using HiRise (v.Aug2019)65. In B. microptera, gaps were closed using TGS-Gapcloser (v.1.1.1)78, haplotigs were removed using Purge Haplotigs (v.1.0.4)79 and the assembly was polished using Illumina WGS reads and pilon (v.1.23)80. In both the sponge and B. microptera genomes, bacterial scaffolds were removed using Diamond (v.0.9.24)81 and Blobtools (v.1.0)82. The genomes were manually curated with Hi-C data as described above. The haplotypes of the hifiasm-based assemblies were compared to one another using D-Genies (v.1.4.0)83.
Genome annotations
The unicellular outgroup genome assemblies were annotated by mapping their transcripts from the original assemblies to the Hi-C scaffolded assemblies using minimap2 (v.2.23)84. To clarify demosponge macrosyntenic relationships, we produced putative Ephydatia protein coordinates in the cladorhizid sponge using tblastn (v.2.10.0+)85. To annotate the hexactinellid sponge genome, we mapped the proteins of closely-related hexactinellid species86,87 using miniprot (v.0.2)88 (Supplementary Information 2.1.5). The Bolinopsis genome was annotated using BRAKER (v.2.14)89 supplied with evidence from RNA-seq reads mapped with STAR (v.2.7.1a)90 and minimap2 (v.2.23)84, Iso-Seq reads processed with lima (v.2.2.0; https://github.com/PacificBiosciences/barcoding) and isoseq3 (v.3.4.0; https://github.com/PacificBiosciences/IsoSeq) then mapped with minimap2 (v.2.23)84, and protein orthology identified using ProtHint (v.2.6.0)91 from ctenophore transcriptomes92,93,94 assembled with Trinity (v.2.5.1)95 and translated using TransDecoder (v.5.5; https://github.com/TransDecoder/TransDecoder). We assessed genome sequence and protein datasets using BUSCO (v.5)96.
Orthologue Inference
Orthologues were inferred between species by finding reciprocal-best BLASTp97 hits between the proteins in the genomes, or with OrthoFinder (v.2.3.7)98. The reciprocal-best BLASTp hits were used to identify macrosyntenic chromosomes between species by performing Bonferroni-corrected one-sided Fisher’s exact tests57. To determine the provenance of the ALG_H in sponges, the genomes of Chondrosia and Petrosia39,99, Oopsacas86, CLA and HEX were compared using the odp software suite.
Orthologues shared between three, four or more species were selected by finding groups of proteins that were n-way reciprocal best BLASTp hits. In this conservative method, each orthogroup has a single protein from each of the n species. We performed this analysis for three-way and four-way comparisons of combinations of the species CFR, COW, SRO, HCA, EMU, CLA, RES, BFL, NVE and P. maximus.
Gene linkage group identification
Orthologues from three-way or four-way reciprocal-best BLASTp searches were grouped by the chromosomes on which the genes occurred in the n species. To identify which sets of orthologues were larger than expected by random chance, we shuffled the genome coordinates of the n species and measured the frequency of finding sets of orthologues of size k on the same chromosomes in the n species. By performing this for 10 million iterations, we calculated the false-discovery rate (α) of finding an orthologue set of size k given the n input genomes.
Combined unicellular outgroup analysis
Sets of orthologues with a false-discovery rate of less than 0.05 were retained from the four-way reciprocal best hit searches of COW–HCA–EMU–RES, CFR–HCA–EMU–RES and SRO–HCA–EMU–RES. The remaining orthogroups were joined based on gene identity in HCA–EMU–RES, such that each orthologue contained a protein from at least one of the unicellular outgroup species. This yielded 291 sets of orthologues.
Identification of orthologues in other species
For each of the 291 orthologues, we aligned the proteins using MAFFT (v.7.310)100, built a hidden Markov Model using hmmbuild in hmmer (v.3.3.2)101, then found the best match using hmmsearch in the proteins of the genomes of other species, including the ctenophore B. microptera, the cladorhizid sponge, T. adhaerens102, H. vulgaris12, N. vectensis103, B. floridae57, P. maximus45 and E. muelleri37. To test for Gene Ontology enrichment of the sets of orthogroups using PANTHER (v.17)104, we also searched for the orthologues in Homo sapiens105.
Mixing analysis
To test whether the _x and _y gene sets present on single chromosomes were well-mixed, we used a metric that counts the number of transitions between a gene in _x to a gene in _y and vice versa. To provide an objective measure of mixing, we computed the α value (false-discovery rate) that the two sets of genes are unmixed by building a distribution of mixing scores from randomly sorted groups of the same size of the _x and _y groups in question. We consider α < 0.05 to be unmixed.
Simulations testing the ctenophore-sister and sponge-sister hypotheses
We applied this methodology to test whether the findings supporting the ctenophore-sister hypothesis were due to the arrangement of any of the observed genomes, implemented as part of the odp software suite. For both the SRO–HCA–EMU–RES and COW–HCA–EMU–RES four-way reciprocal best hit results, we performed four analyses. One analysis shuffles the genome chromosome labels of one species 100 million times. Each time the genome chromosome labels are shuffled, we perform the gene linkage group identification analysis described above, and measure the quantity and size of gene linkage groups that support either the ctenophore-sister or sponge-sister hypothesis. The distribution of these results compared with the observed data of the real genomes is used to estimate the false-discovery rate of finding support for the ctenophore-sister hypothesis or sponge-sister hypothesis. We modelled fusion-with-mixing events in the animal genomes as state transitions, and used RevBayes (v.1.1.1)106 and MrBayes (v.3.2.7a)52 to estimate the likelihood of the ctenophore-sister hypothesis, and we used FigTree (v.1.4.4; https://github.com/rambaut/figtree) to visualize the trees.
Software
We implemented a suite of tools for identifying orthologues, plotting syntenic relationships and performing synteny-based phylogenetic analyses using a tool called odp, implemented in snakemake (v.7)107 for scalability. To confirm the validity of these methods, we applied them to several genome quartets and showed that odp recovers previously identified synapomorphic chromosomal fusion-with-mixing events12 in bilaterians and cnidarians (Supplementary Information 6).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data presented in this Article are available in public repositories. The sequencing reads are available in the NCBI database under BioProject accession numbers PRJNA818620, PRJNA818630, PRJNA903214 and PRJNA818537. The genomes for each species are available through the above BioProject accession codes, with the exception of the genomes of C. fragrantissima, C. owczarzaki and S. rosetta, which are available at Dryad (https://doi.org/10.5061/dryad.dncjsxm47). The results shown in the Supplementary Information, when not contained in figures, are also available in the Dryad repository (https://doi.org/10.5061/dryad.dncjsxm47). Publicly available sequencing data and genomes were downloaded from NCBI from BioProject accession numbers PRJNA168, PRJDB8655, PRJNA12874, PRJNA20249, PRJNA20341, PRJEB28334, PRJNA30931, PRJNA31257, PRJNA37927, PRJEB56075, PRJEB56892, PRJNA64405, PRJNA193541, PRJNA193613, PRJNA213480, PRJNA278284, PRJNA281977, PRJNA283290, PRJNA377365, PRJNA396415, PRJNA512552, PRJNA544471, PRJNA576068, PRJNA579531, PRJNA625562, PRJNA667495, PRJNA761294 and PRJNA814716. The E. muelleri genome was downloaded from https://spaces.facsci.ualberta.ca/ephybase/.
Code availability
The scripts and software developed for this manuscript are available at Dryad (https://doi.org/10.5061/dryad.dncjsxm47) and Zenodo (https://doi.org/10.5281/zenodo.7857390). Long-term development of odp is available at GitHub (https://github.com/conchoecia/odp).
References
Ryan, J. F. et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342, 1242592 (2013).
Halanych, K. M. The ctenophore lineage is older than sponges? That cannot be right! Or can it? J. Exp. Biol. 218, 592–597 (2015).
Jékely, G., Paps, J. & Nielsen, C. The phylogenetic position of ctenophores and the origin(s) of nervous systems. Evodevo 6, 1 (2015).
Ryan, J. F. & Chiodin, M. Where is my mind? How sponges and placozoans may have lost neural cell types. Philos. Trans. R. Soc. Lond. B 370, 20150059 (2015).
Moroz, L. L. et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 510, 109–114 (2014).
Burkhardt, P. Ctenophores and the evolutionary origin(s) of neurons. Trends Neurosci. 45, 878–880 (2022).
Philippe, H. et al. Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 19, 706–712 (2009).
Simion, P. et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 27, 958–967 (2017).
Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008).
Whelan, N. V. et al. Ctenophore relationships and their placement as the sister group to all other animals. Nat. Ecol. Evol. 1, 1737–1746 (2017).
Li, Y., Shen, X.-X., Evans, B., Dunn, C. W. & Rokas, A. Rooting the animal tree of life. Mol. Biol. Evol. 38, 4322–4333 (2021).
Simakov, O. et al. Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 8, eabi5884 (2022).
Dunn, C. W., Leys, S. P. & Haddock, S. H. D. The hidden biology of sponges and ctenophores. Trends Ecol. Evol. 30, 282–291 (2015).
Ryan, J. F. et al. The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. Evodevo 1, 9 (2010).
Musser, J. M. et al. Profiling cellular diversity in sponges informs animal cell type and nervous system evolution. Science 374, 717–723 (2021).
Harbison, G. R. in The Origins and Relationships of Lower Invertebrates Systematics Association Special Vol. 28 (eds Morris, S. C. et al.) 78–100 (Clarendon Press, 1985).
Tamm, S. L. Formation of the statolith in the ctenophore Mnemiopsis leidyi. Biol. Bull. 227, 7–18 (2014).
Burton, P. M. Insights from diploblasts; the evolution of mesoderm and muscle. J. Exp. Zool. B 310, 5–14 (2008).
Sachkova, M. Y. et al. Neuropeptide repertoire and 3D anatomy of the ctenophore nervous system. Curr. Biol. 31, 5274–5285 (2021).
Antcliffe, J. B., Callow, R. H. T. & Brasier, M. D. Giving the early fossil record of sponges a squeeze. Biol. Rev. Camb. Philos. Soc. 89, 972–1004 (2014).
O’Brien, L. J. & Caron, J.-B. A new stalked filter-feeder from the middle Cambrian Burgess Shale, British Columbia, Canada. PLoS ONE 7, e29233 (2012).
Mah, J. L. & Leys, S. P. Think like a sponge: the genetic signal of sensory cells in sponges. Dev. Biol. 431, 93–100 (2017).
Erives, A. & Fritzsch, B. A screen for gene paralogies delineating evolutionary branching order of early metazoa. G3 10, 811–826 (2020).
Shen, X.-X., Hittinger, C. T., Rokas, A., Minh, B. Q. & Braun, E. L. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 1, 0126 (2017).
Francis, W. R. & Canfield, D. E. Very few sites can reshape the inferred phylogenetic tree. PeerJ 8, e8865 (2020).
Kapli, P., Yang, Z. & Telford, M. J. Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 21, 428–444 (2020).
Renwick, J. H. The mapping of human chromosomes. Annu. Rev. Genet. 5, 81–120 (1971).
Muller, H. J. in The New Systematics (ed. Huxley, J.) 185–268 (Oxford Univ. Press, 1940).
Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450, 203–218 (2007).
Hane, J. K. et al. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 12, R45 (2011).
Wright, S. On the probability of fixation of reciprocal translocations. Am. Nat. 75, 513–522 (1941).
Lv, J., Havlak, P. & Putnam, N. H. Constraints on genes shape long-term conservation of macro-synteny in metazoan genomes. BMC Bioinform. 12, S11 (2011).
Hillis, D. M. SINEs of the perfect character. Proc. Natl Acad. Sci. USA 96, 9979–9981 (1999).
Rokas, A. & Holland, P. W. Rare genomic changes as a tool for phylogenetics. Trends Ecol. Evol. 15, 454–459 (2000).
Okada, N. SINEs: Short interspersed repeated elements of the eukaryotic genome. Trends Ecol. Evol. 6, 358–361 (1991).
Martini, S., Schultz, D. T., Lundsten, L. & Haddock, S. H. D. Bioluminescence in an undescribed species of carnivorous sponge (Cladorhizidae) from the deep sea. Front. Mar. Sci. 7, 576476 (2020).
Kenny, N. J. et al. Tracing animal genomic evolution with the chromosomal-level assembly of the freshwater sponge Ephydatia muelleri. Nat. Commun. 11, 3676 (2020).
Schuster, A. et al. Divergence times in demosponges (Porifera): first insights from new mitogenomes and the inclusion of fossils in a birth-death clock model. BMC Evol. Biol. 18, 114 (2018).
McKenna, V. et al. The Aquatic Symbiosis Genomics Project: probing the evolution of symbiosis across the tree of life. Wellcome Open Res. 6, 254 (2021).
Schultz, D. T. et al. A chromosome-scale genome assembly and karyotype of the ctenophore Hormiphora californensis. G3 11, jkab302 (2021).
Johnson, S. B. et al. Speciation of pelagic zooplankton: invisible boundaries can drive isolation of oceanic ctenophores. Front. Genet. 13, 970314 (2022).
Fairclough, S. R. et al. Premetazoan genome evolution and the regulation of cell differentiation in the choanoflagellate Salpingoeca rosetta. Genome Biol. 14, R15 (2013).
Suga, H. et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nat. Commun. 4, 2325 (2013).
de Mendoza, A., Suga, H., Permanyer, J., Irimia, M. & Ruiz-Trillo, I. Complex transcriptional regulation and independent evolution of fungal-like traits in a relative of animals. eLife 4, e08904 (2015).
Kenny, N. J. et al. The gene-rich genome of the scallop Pecten maximus. Gigascience 9, giaa037 (2020).
Li, Y. et al. Chromosome-level reference genome of the jellyfish Rhopilema esculentum. Gigascience 9, giaa036 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Lang, B. F., O’Kelly, C., Nerad, T., Gray, M. W. & Burger, G. The closest unicellular relatives of animals. Curr. Biol. 12, 1773–1778 (2002).
Torruella, G. et al. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol. Biol. Evol. 29, 531–544 (2012).
Strassert, J. F. H., Irisarri, I., Williams, T. A. & Burki, F. A molecular timescale for eukaryote evolution with implications for the origin of red algal-derived plastids. Nat. Commun. 12, 1879 (2021).
Schubert, I. & Lysak, M. A. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 27, 207–216 (2011).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Hyman, L. H. The phylogeny of the lower metazoa. Q. Rev. Biol. 51, 344–347 (1976).
Collins, A. G. Evaluating multiple alternative hypotheses for the origin of Bilateria: an analysis of 18S rRNA molecular evidence. Proc. Natl Acad. Sci. USA 95, 15458–15463 (1998).
Laumer, C. E. et al. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. Biol. Sci. 286, 20190831 (2019).
Schierwater, B. My favorite animal, Trichoplax adhaerens. Bioessays 27, 1294–1302 (2005).
Simakov, O. et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830 (2020).
Fernández, R. & Gabaldón, T. Gene gain and loss across the metazoan tree of life. Nat. Ecol. Evol. 4, 524–533 (2020).
Moroz, L. L. Multiple origins of neurons from secretory cells. Front. Cell Dev. Biol. 9, 669087 (2021).
Belahbib, H. et al. New genomic data and analyses challenge the traditional vision of animal epithelium evolution. BMC Genom. 19, 393 (2018).
Albalat, R. & Cañestro, C. Evolution by gene loss. Nat. Rev. Genet. 17, 379–391 (2016).
Parker, G. H. The Elementary Nervous System (J.B. Lippincott Company, 1919).
Leys, S. P. Elements of a ‘nervous system’ in sponges. J. Exp. Biol. 218, 581–591 (2015).
Adams, M. et al. One fly-one genome: chromosome-scale genome assembly of a single outbred Drosophila melanogaster. Nucleic Acids Res. 48, e75 (2020).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016).
Ghurye, J., Pop, M., Koren, S., Bickhart, D. & Chin, C.-S. Scaffolding of long read assemblies using long range contact information. BMC Genom. 18, 527 (2017).
Kerpedjiev, P. et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 19, 125 (2018).
Dudchenko, O. et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Preprint at bioRxiv https://doi.org/10.1101/254797 (2018).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Denbo, S. et al. Revision of the Capsaspora genome using read mating information adjusts the view on premetazoan genome. Dev. Growth Differ. 61, 34–42 (2019).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Open2C et al. Pairtools: from sequencing data to chromosome contacts. Preprint at bioRxiv https://doi.org/10.1101/2023.02.13.528389 (2023).
Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316 (2020).
Xu, M. et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 9, giaa094 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 460 (2018).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Laetsch, D. R. & Blaxter, M. L. BlobTools: interrogation of genome assemblies. F1000Res. 6, 1287 (2017).
Cabanettes, F. & Klopp, C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6, e4958 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Gertz, E. M., Yu, Y.-K., Agarwala, R., Schäffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4, 41 (2006).
Santini, S. et al. The compact genome of the sponge Oopsacas minuta (Hexactinellida) is lacking key metazoan core genes. Preprint at bioRxiv https://doi.org/10.1101/2022.07.26.501511 (2022).
Whelan, N. V., Kocot, K. M., Moroz, L. L. & Halanych, K. M. Error, signal, and the placement of Ctenophora sister to all other animals. Proc. Natl Acad. Sci. USA 112, 5773–5778 (2015).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-genome annotation with BRAKER. Methods Mol. Biol. 1962, 65–95 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2, lqaa026 (2020).
Francis, W. R., Shaner, N. C., Christianson, L. M., Powers, M. L. & Haddock, S. H. D. Occurrence of isopenicillin-N-synthase homologs in bioluminescent ctenophores and implications for coelenterazine biosynthesis. PLoS ONE 10, e0128742 (2015).
Townsend, J. P. et al. A mesopelagic ctenophore representing a new family, with notes on family-level taxonomy in Ctenophora: Vampyroctena delmarvensis gen. nov. sp. nov. (Vampyroctenidae, fam. nov.). Mar. Biodivers. 50, 34 (2020).
Babonis, L. S. et al. Integrating embryonic development and evolutionary history to characterize tentacle-specific cell types in a ctenophore. Mol. Biol. Evol. 35, 2940–2956 (2018).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
The Darwin Tree of Life Project. Sequence locally, think globally: The Darwin Tree of Life Project. Proc. Natl Acad. Sci. USA 119, e2115642118 (2022).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
Srivastava, M. et al. The Trichoplax genome and the nature of placozoans. Nature 454, 955–960 (2008).
Zimmermann, B. et al. Sea anemone genomes reveal ancestral metazoan chromosomal macrosynteny. Preprint at bioRxiv https://doi.org/10.1101/2020.10.30.359448 (2020).
Mi, H., Muruganujan, A., Casagrande, J. T. & Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566 (2013).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Höhna, S. et al. RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model-specification language. Syst. Biol. 65, 726–736 (2016).
Köster, J. & Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012).
Acknowledgements
We acknowledge the support of the David and Lucile Packard Foundation, the Monterey Bay Aquarium Research Institute and the United States National Science Foundation (NSF). D.T.S. was supported by the NSF GRFP DGE 1339067; S.H.D.H. by NSF DEB-1542679; D.T.S. and O.S. by the European Research Council’s Horizon 2020: European Union Research and Innovation Programme, grant no. 945026; D.S.R. by internal funds of the OIST Molecular Genetics Unit, the Chan Zuckerberg Biohub and the Marthella Foskett Brown Chair in Biology. We thank W. Patry and the staff at Monterey Bay Aquarium for providing the Bolinopsis specimen; and C. Dunn, J. Eizenga, R. Revilla-i-Domingo and G. Genikhovich for discussions. The Petrosia and Chondrosia genomes from the Aquatic Symbiosis Genomics project of the Tree of Life Programme, Wellcome Sanger Institute, were funded by the Gordon and Betty Moore Foundation and the Wellcome Trust.
Author information
Authors and Affiliations
Contributions
D.T.S., O.S. and D.S.R. designed the scientific objectives of the study. D.T.S. planned and carried out experiments and analyses, wrote the code for the analyses and the odp package, and wrote the first draft of the manuscript. O.S. and D.T.S. performed the Bayesian analyses. D.T.S. and S.H.D.H. collected samples and created figures and tables. J.V.B. assisted in genome assemblies and identified putative centromeres. D.T.S. and D.S.R. wrote the first drafts of the manuscript with input from O.S. and S.H.D.H. All of the authors contributed to the interpretation, presentation and writing of the Article and the Supplementary Information.
Corresponding authors
Ethics declarations
Competing interests
R.E.G. and D.S.R. are paid consultants and equity holders of Dovetail Genomics. D.T.S. is a shareholder of Pacific Biosciences of California. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Genomes of one ctenophore and two sponges.
a. The k-mer spectrum of the Bolinopsis data suggests that the animal is diploid, and the 1n genome size is approximately 254 Mbp. b. The Bolinopsis microptera genome assembly contains 13 chromosome-scale scaffolds, which account for 97.23% of the total bases in the assembly. Panel shows the Hi-C contact map. c. The cladorhizid sponge individual used in the genome sequencing at its collection site. d. This sponge was bioluminescent when mechanically disturbed. e. Its mitochondrial sequence is 99.2% identical to the previously identified bioluminescent cladorhizid sponge36. f. The estimated genome size of this sponge is 1.11 Gb, and the spectrum is consistent with diploid organisms. g.,h. Each haplotype’s genome assembly has 18 chromosome-scale scaffolds based on chromatin confirmation data as shown. In haplotype A 94.2% of bases are in the chromosome-scale scaffolds. i. A whole-genome alignment of haplotypes A and B showed a high degree of concordance. j. The hexactinellid sponge collected and sequenced for this study. k. The estimated genome size is 1n = 141 Mb. l. Haplotype A contains only one haplotype of chromosome-scale scaffolds orthologous with the scaffolds of the closely-related sponge Oopsacas minuta. Panel shows chromatin conformation capture contact map of haplotype A. m. In addition to the alternate haplotype of chromosome-scale scaffolds from haplotype A, the haplotype B assembly contains the large, gene-poor, unplaced scaffolds that lack detectable homology to other sponges. The Hi-C contact map for haplotype B shown. n. Whole-genome alignments of the two haplotypes show colinearity. Photograph credits: (d.) Darrin Schultz, (c., j.) © 2021 MBARI.
Extended Data Fig. 2 Chromosomes are largely conserved among metazoans.
The chromosome position of orthologous proteins plotted in panels a-n are coloured by orthologs in the previously identified ancestral bilaterian, cnidarian, and sponge linkage groups (BCnS-ALGs)12. Significant inter-species chromosome pairs (p ≤ 0.05, Bonferroni-corrected one-sided Fisher’s exact test57) are opaque. a. The karyotype of the Pleurobrachiid and lobate ctenophores is conserved (1n = 13). b.-e. Ctenophore chromosomes share macrosynteny with BCnS-ALGs, but many BCnS ALGs are split onto several ctenophore chromosomes (red dotted boxes). There are many ctenophore-specific chromosome fusions. f.-h. Macrosynteny is highly conserved between distantly-related demosponges. The sponge lineages shown diverged an estimated 358 Mya - 500 Mya38. f.-k. Macrosynteny is also conserved between sponge, bilaterian, and cnidarian genomes. Many chromosomes in a species of one clade have a one-to-one homologous chromosome in the other clade. The genomes of species in these clades can be described by 29 constituent BCnS-ALGs12.
Extended Data Fig. 3 Sponge macrosynteny.
a.-c. There have been many genome rearrangements since the divergence of the demosponge Ephydatia and the tulip hexactinellid genome, and they share macrosynteny of only some BCnS linkage groups (p ≤ 0.05, Bonferroni-corrected one-sided Fisher’s exact test57, opaque dots in a., rows in b., interspecies lines in c.). d. The sponge cladogram is based on Schuster et al. 201838. e. The orthologs in A1a_x and A1a_y are predominantly present on separate chromosomes in both the tulip hexactinellid and in Oopsacas minuta. f. A1a_x and A1a_y are on partly overlapping regions of single demosponge chromosomes, but are mixed on a single Chondrosia chromosome. However, the linkage groups A1a_x and A1a_y are on separate chromosomes in the ctenophores and the unicellular outgroup species. This evidence suggests that hexactinellid sponges retain the ancestral state of A1a_x and A1a_y being present on separate chromosomes. The possible evolutionary scenarios explaining this karyotype will require further chromosome-scale sequencing of sponge genomes.
Extended Data Fig. 4 Seven basic ALG configurations in species quartets.
a-g. The seven configurations of ALGs found in four species highlight the evolutionary history of chromosomes. The cartoon ribbon plot in each panel shows chromosomes (horizontal bars) and the positions of genes in two ALGs along those chromosomes (vertical blue or red lines, respectively). The cartoon Oxford dot plot in each panel shows the same information as the ribbon plot, but only in the context of the outgroup genome. The most parsimonious tree topology based on the ALG evolutionary history is also pictured.
Extended Data Fig. 5 Unicellular species chromosome-scale genome assemblies.
Hi-C heatmaps of a. Salpingoeca rosetta, b. Capsaspora owczarzaki, and c. Creolimax fragrantissima show that the assemblies are consistent with chromosome-scale assemblies of other unicellular species. d.-e. Genome-wide ICE-normalized 108 observed count contact maps for (d.) Salpingoeca, (e.) Capsaspora, and (f.) Creolimax are shown at MapQ0 and 10 kb resolution. Chromosome boundaries are drawn as solid black lines. The intersections of horizontal and vertical red lines mark the Centurion-estimated centromere positions. The Hi-C heatmaps of Capsaspora and Creolimax both contain inter-chromosomal hotspots that are consistent with centromeres in other species. g.-i. Protein orthology plots (Oxford dot plots) of the chromosome-scale genome assemblies compared to the previously published assemblies. Despite the lack of Hi-C data, the original scaffold assemblies for all three species were nearly chromosome-scale.
Extended Data Fig. 6 Visual representation of multi-species gene linkage conservation score.
a.-d. The dot plots of the C. owczarzaki genome show that there is conservation of the ALG_A1a linkage group in ctenophore, sponge, cnidarian, and bilaterian genomes. The conservation score can be calculated from shared gene linkages across many species. f. Due to the highly rearranged state of both the Hormiphora and Capsaspora genomes, a Bonferroni-corrected one-sided Fisher’s exact test57 only distinguishes three chromosome relationships as significant (p ≤ 0.05). e. Calculating the orthology conservation score for the relationships in these two genomes reveals more gene linkages that have been conserved across Filozoans. Red dots here are orthologs that are in significantly-conserved ortholog networks (α ≤ 0.05, permutation test). See complete results in Supplementary Information 11.
Extended Data Fig. 7 Filozoan and choanoflagellate genomes share macrosynteny with metazoans.
Two-way reciprocal best hits blast searches between the filasterean amoeba Capsaspora and animals (a.-d.), or between the choanoflagellate Salpingoeca and animals (d.-g.) show that the chromosomes of these unicellular species are rearranged relative to animal chromosomes, that some regions of synteny remain, and that some ALGs are split across multiple chromosomes of the unicellular species. Orthologs are coloured based on BCnS-ALGs from Simakov et al. 202212, and chromosome pairs with significantly-conserved macrosynteny (p ≤ 0.05, Bonferroni-corrected one-sided Fisher’s exact test57) have opaque dots. Axis labels show cumulative number of orthologs. Putative centromeres are marked by dotted lines.
Extended Data Fig. 8 Mixing plots of HCA-EMU-RES reciprocal best blastp results.
This figure parallels Fig. 3 of the main text, but includes more genes by requiring orthology between metazoans without requiring orthologs in corresponding outgroups. Limiting the macrosynteny search to animals shows many genes participating in the extension of metazoan ALGs to the ctenophores. The _x and _y components of ALG_Ea and ALG_G are mixed and widely distributed across single sponge chromosomes, while the (COW/SRO)-HCA-EMU-RES results show no _x and _y overlap for ALG_Ea, and little overlap for ALG_G. We placed placozoans as the sister clade to cnidarians based on the findings of Simakov et al. 202212. See also Supplementary Information 13.2.2.
Extended Data Fig. 9 OrthoFinder results are consistent with the ctenophore-sister hypothesis.
a. Each green cell shows how many orthogroups support the ctenophore-sister hypothesis from each ALG in each species quartet. The Total Gene Count column is the total number of orthogroups supporting the ctenophore-sister hypothesis for that species quartet. The bottom row shows the number of unique orthogroups in each column. There are 146 orthogroups that support ctenophore-sister. b. The 11 orthologs that support CLA-sister in three analyses are due to a lineage-specific fission of ALG_H that is only found in the cladorhizid genome, but not in the genome of other sponges. Tree topology based on previous studies38,109. c. The Capsaspora-cladorhizid chromosome pairs with the most genes from ALG_H (COW4-CLA13, COW4-CLA14) are not the chromosome pairs supporting sponge-sister (magenta circles, COW4-CLA13, COW6-CLA14). d.-h. The fission of ALG_H is specific to the cladorhizid sponge genome and is not found in the unicellular organism Capsaspora (COW), in other demosponges (EMU, CRE, PFI), in cnidarians (RES), or in bilaterians (not shown). Chromosome pairs that have significantly-conserved macrosynteny (p ≤ 0.05, Bonferroni-corrected one-sided Fisher’s exact test57) have opaque dots.
Extended Data Fig. 10 Results of genome shuffling simulations.
a.-d. Shuffling one of the genomes before the COW-HCA-EMU-RES comparison shows that the rearranged state of the ctenophore genome, let alone the other species in the analysis, cannot explain the signal supporting the ctenophore-sister hypothesis (vertical red lines). e.-h. Shuffling simulations using SRO as the outgroup independently support the ctenophore-sister hypothesis. i. contains a legend to interpret panels a-h.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary Tables, Supplementary Figures, Supplementary Results and Discussion, and Supplementary Notes.
Supplementary Data 1
Sequencing library details.
Supplementary Data 2
Orthologue tables.
Supplementary Data 3
GO analysis.
Supplementary Data 4
Text files of ALG mixing results.
Supplementary Data 5
Tables of ALG mixing results.
Supplementary Data 6
Bayesian analysis files.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schultz, D.T., Haddock, S.H.D., Bredeson, J.V. et al. Ancient gene linkages support ctenophores as sister to other animals. Nature 618, 110–117 (2023). https://doi.org/10.1038/s41586-023-05936-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-05936-6
This article is cited by
-
Neuromuscular organization of the benthic ctenophore, Vallicula multiformis
Zoological Letters (2024)
-
The ctenophore Mnemiopsis leidyi deploys a rapid injury response dating back to the last common animal ancestor
Communications Biology (2024)
-
Extracellular DNA traps in a ctenophore demonstrate immune cell behaviors in a non-bilaterian
Nature Communications (2024)
-
The origin and evolution of Wnt signalling
Nature Reviews Genetics (2024)
-
Telomeres and telomerase: active but complex players in life-history decisions
Biogerontology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
