
Abstract
Acinic cell carcinoma is an indolent form of invasive breast cancer, whereas microglandular adenosis has been shown to be a neoplastic proliferation. Both entities display a triple-negative phenotype, and may give rise to and display somatic genomic alterations typical of high-grade triple-negative breast cancers. Here we report on a comparison of previously published data on eight carcinoma-associated microglandular adenosis and eight acinic cell carcinomas subjected to targeted massively parallel sequencing targeting all exons of 236 genes recurrently mutated in breast cancer and/or DNA repair-related. Somatic mutations, insertions/ deletions, and copy number alterations were detected using state-of-the-art bioinformatic algorithms. All cases were of triple-negative phenotype. A median of 4.5 (1–13) and 4.0 (1–7) non-synonymous somatic mutations per carcinoma-associated microglandular adenosis and acinic cell carcinoma were identified, respectively. TP53 was the sole highly recurrently mutated gene (75% in microglandular adenosis versus 88% in acinic cell carcinomas), and TP53 mutations were consistently coupled with loss of heterozygosity of the wild-type allele. Additional somatic mutations shared by both groups included those in BRCA1, PIK3CA, and INPP4B. Recurrent (n=2) somatic mutations restricted to microglandular adenosis or acinic cell carcinomas included those affecting PTEN and MED12 or ERBB4, respectively. No significant differences in the repertoire of somatic mutations were detected between microglandular adenosis and acinic cell carcinomas, and between this group of lesions and 77 triple-negative carcinomas from The Cancer Genome Atlas. Microglandular adenosis and acinic cell carcinomas, however, were genetically distinct from estrogen receptor-positive and/or HER2-positive breast cancers from The Cancer Genome Atlas. Our findings support the contention that microglandular adenosis and acinic cell carcinoma are part of the same spectrum of lesions harboring frequent TP53 somatic mutations, and likely represent low-grade forms of triple-negative disease with no/minimal metastatic potential, of which a subset has the potential to progress to high-grade triple-negative breast cancer.
Similar content being viewed by others


Main
Microglandular adenosis of the breast encompasses a spectrum of lesions, ranging from pure forms without atypia, to atypical microglandular adenosis and carcinoma-associated lesions.1 Despite being historically named ‘adenosis’, several studies have reported on the progression from microglandular adenosis /atypical microglandular adenosis to invasive carcinomas, which are mostly of high histologic grade and triple-negative immunophenotype (ie, lacking expression of estrogen receptor (ER), progesterone receptor (PR), and HER2).2, 3, 4, 5 In fact, molecular analyses have suggested that microglandular adenoses /atypical microglandular adenoses, at least those associated with carcinoma, are clonal neoplastic lesions and non-obligate precursors of high-grade triple-negative breast cancers, as synchronously diagnosed ipsilateral microglandular adenoses/atypical microglandular adenoses and invasive carcinomas display similar patterns of copy number alterations and mutation profiles.6, 7, 8, 9, 10
Acinic cell carcinoma is a rare special histologic type of breast cancer of low-grade and indolent behavior,11 which, akin to microglandular adenoses/atypical microglandular adenoses, may progress to high-grade triple-negative breast cancer.12, 13 Indeed, metastatic potential may be limited to those cases mixed with a high-grade/non-acinic cell component.11 Despite these favorable prognostic features, acinic cell carcinomas display complex patterns of copy number alterations and harbor highly recurrent TP53 mutations,12 paralleling the genomic profiles of common forms of triple-negative breast cancers.14
Despite conceptual differences, microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas display histological and immunohistochemical similarities.1, 11, 13, 15 Morphologically, both entities typically are characterized by an infiltrative proliferation of small glands lined by low-grade cuboidal to flattened cells lacking a myoepithelial cell layer.1, 11 Acinic cell carcinomas may however display a distinct architecture such as the hypernephroid clear cell pattern,12 and may show intra-tumor heterogeneity with well-differentiated tubular and less-differentiated solid areas.13 Immunophenotypically, microglandular adenoses/atypical microglandular adenoses, and acinic cell carcinomas are characterized by strong expression of S100 protein4, 5, 6, 7, 9, 16 and most are of triple-negative immunophenotype.4, 6, 7, 9, 12, 13, 16 Differential diagnosis of microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas can be challenging15, 16 and often relies on the identification of diffuse serous differentiation in acinic cell carcinomas.11, 16 The latter can be defined by the presence of intracytoplasmic zymogen-type granules or expression of acinar differentiation markers, such as lysozyme and amylase. Microglandular adenosis/atypical microglandular adenosis cells, however, have been shown to focally display these features.4, 5, 15
Large-scale genomic studies have been conducted in breast cancer, demonstrating that only three genes are mutated in more than 10% of unselected cases, namely TP53 (37%), PIK3CA (36%), and GATA3 (11%).14, 17 In the subset of triple-negative breast cancers, even greater inter-tumor heterogeneity is observed. Only TP53 is highly recurrently mutated (86%), whereas in TP53 wild-type cancers, the p53 pathway is usually inactivated by other mechanisms.14, 17 In addition, triple-negative breast cancers are characterized by the loss of cell cycle checkpoints RB1 and BRCA1, and by activation of the PI3K pathway through several mechanisms including PIK3CA mutations (9%) or amplifications, and loss of PTEN or INPP4B.14, 17, 18
Given the histological and immunohistochemical similarities between microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas, and between this group of lesions and triple-negative breast cancers, and the observation that microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas may progress to high-grade triple-negative breast cancers, here we sought to compare the genomic landscape of microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas and to define the similarities and differences between these lesions and high-grade triple-negative breast cancers.
Patients and methods
Cases and Immunohistochemistry
All cases included in this study have been previously described.9, 12 Here we report on a re-analysis of previously published targeted massively parallel sequencing data reported by Guerini-Rocco et al.9 and Guerini-Rocco et al.12 including 10 microglandular adenoses and/or microglandular atypical microglandular adenoses, of which eight were associated with in situ and/or invasive carcinomas, and eight cases of acinic cell carcinoma, of which six were admixed with non-acinic cell carcinoma components (Table 1). All cases were microdissected (tumor and paired histologically normal breast tissue). The immunohistochemical features of cases included in this study have been previously described,9, 12 apart from immunohistochemical analysis using antibodies against lysozyme in three MGA cases (MGA4, MGA5, and MGA8), which was performed as previously described,12 with a polyclonal anti-lysozyme antibody (Ventana) using the CC1 mild pretreatment program on a Benchmark XT autostainer (Ventana). Lysozyme was considered positive if cytoplasmic staining was observed in >1% of tumor cells.19 Positive and negative controls were included in each slide run.
Targeted Capture Massively Parallel Sequencing
The massively parallel sequencing data were retrieved from SRA (accession SRP062955 and SRP052551) and analyzed essentially as previously described20 using state-of-the-art algorithms.21, 22, 23, 24, 25 Experiments were previously performed using customized sequencing assays targeting all exons of 254,12 273 (ref. 9) or 297 (ref. 9) genes recurrently mutated in breast cancer, genes related to DNA repair and/ or potentially actionable cancer genes, with 236 genes present on all three platforms (Supplementary Table S1). For those cases with microglandular adenosis and atypical microglandular adenosis components (MGA7 and MGA13) or those acinic cell carcinomas with acinic cell components of distinct histologic grades (1 and 2) and/or distinct growth patterns (microglandular and clear cell; ACC14 and ACC16), we merged the raw data of these samples before the comparison. Of note, the morphologically distinct components within these cases displayed almost identical genetic profiles.9, 12 The functional effect of each missense single-nucleotide variant was investigated as previously described.26, 27, 28 Genes affected by non-passenger mutations were assessed for their presence in three cancer gene datasets, Kandoth et al.,29 the Cancer Gene Census30 and Lawrence et al.31 Allele-specific copy number alterations were identified using FACETS as previously described.32, 33 In brief, positions within the target regions with dbSNP entries (build 137) were retrieved and read counts were generated for matched tumor and normal samples. These read counts were employed as input to FACETS, which performs a joint segmentation of the total and allelic copy ratios. Genes with copy number alterations were identified by adopting the approaches described in Curtis et al.33 and elsewhere.9, 20 In brief, the median log2 ratio +2 standard deviations (s.d.’s) or +6s.d. was computed for the 50% (or 45% or 40%, see below) of the central positions ordered by their log2 ratios to call copy number gains and amplifications, respectively, and the median log2 ratio −2.5s.d.’s or −7s.d.’s was computed for the 50% (or 45% or 40%) of the central positions to call copy number losses and homozygous deletions, respectively.9, 20 To account for the differences in tumor cell content, ploidy and noise between the samples, the proportion of central positions, ordered by their log2 ratios, was determined based on the median absolute difference between the raw log2 ratio and the segmented log2 ratio. For samples for which the median absolute difference was ≤0.2, >0.2 and ≤0.3 and >0.3, 50%, 45%, and 40%, respectively, of the central positions were used. Lesser copy number estimates of each segment were used to determine whether genes harboring a somatic mutation were targeted by loss of heterozygosity. The output generated by FACETS was subsequently reviewed, and all gene amplifications, homozygous deletions and loss of heterozygosity were visually inspected using plots of raw log2 and allele ratios. To define the cancer cell fraction of each mutation, ABSOLUTE (v1.0.6)34 was employed, based on the number of reads supporting the reference and the alternate alleles and the segmented log2 ratio from targeted capture massively parallel sequencing as input. Solutions from ABSOLUTE were manually reviewed as recommended.34, 35 A mutation was classified as clonal if its probability of being clonal was >50%35 or if the lower bound of the 95% confidence interval of its cancer cell fraction was >90%. Mutations that did not meet the above criteria were considered subclonal.
Statistical Analysis and Comparisons with Triple-Negative Breast Cancers from the The Cancer Genome Atlas Data Set
A comparison of overall mutation rate was performed using Mann–Whitney U test, whereas the comparisons of mutation rates affecting single genes and copy number alterations were performed using Fisher’s exact tests. To compare the genomic profiles of carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas to those of triple-negative, ER-positive/HER2-negative and HER2-positive breast cancers from The Cancer Genome Atlas study of breast cancers, we retrieved the clinico-pathological data and whole-exome sequencing-derived mutational and copy number data from The Cancer Genome Atlas data portal (https://tcga-data.nci.nih.gov/docs/publications/brca_2012/; files ‘Key Clinical Data’, ‘UCEC Somatic Mutations’, ‘Cumulative Data Freeze List’). We restricted the comparison of the genomic features of our cohort of microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas to those of invasive breast cancers annotated as ER-negative/PR-negative/HER2-negative (N=77), ER-positive/HER2-negative (N=252), and HER2-positive (N=91; defined as 3+ by immunohistochemistry or amplified by in situ hybridization). In this analysis, only the 236 genes included in the targeted capture massively parallel sequencing assays used in this study were analyzed (Supplementary Table S1).
Results
Microglandular Adenoses/Atypical Microglandular Adenoses and Acinic Cell Carcinomas are of Triple-Negative Phenotype and Express Lysozyme
The clinical, histological and immunohistochemical features of microglandular adenosis/atypical microglandular adenosis and acinic cell carcinoma cases included in this study have been previously described9, 12 and are summarized in Table 1 and Figure 1. Microglandular adenosis cases included two pure microglandular adenoses (ie, not associated with atypia or carcinoma) and eight carcinoma-associated microglandular adenoses/atypical microglandular adenoses. Acinic cell carcinoma cases included two pure and six cases where a non-acinic cell component was synchronously present. Five and three acinic cell components of acinic cell carcinomas were of histologic grades 1 and 2, respectively. All but one microglandular adenosis-associated invasive carcinomas and all but one non-acinic cell components of mixed acinic cell carcinomas were of histologic grade 3. Notably, all components of all cases were of triple-negative phenotype. All microglandular adenoses, atypical microglandular adenoses, and microglandular adenosis-associated carcinomas tested for S100 were positive.9 All acinic-cell components of acinic cell carcinomas and three non-acinic cell components of six mixed acinic cell carcinomas were positive for lysozyme.12 Owing to the morphologic similarities between microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas, we aimed to investigate whether microglandular adenoses/atypical microglandular adenoses would also display immunohistochemical serous differentiation, using the same protocol as that used for the immunohistochemical analysis of acinic cell carcinomas in our previous study.12 Available tissue was retrieved from cases MGA4, MGA5, and MGA8; in these cases lysozyme expression was observed in both microglandular adenosis and invasive carcinoma components (Figure 1). In two cases, lysozyme was diffusely and intensely expressed, whereas in the third case, its expression was focal.



Representative micrographs of histological features of and the expression of lysozyme expression in microglandular adenoses and acinic cell carcinomas. (a and b) Carcinoma-associated microglandular adenosis. (c and d) Microglandular adenosis-associated invasive carcinoma. (e and f) Pure acinic cell carcinoma. (g and h) Mixed acinic cell carcinoma, with acinic cell component on the right and non-acinic cell component on the left. Please note lysozyme expression in all lesions. Original magnification 200 ×.
Carcinoma-Associated Microglandular Adenoses/ Atypical Microglandular Adenoses and Acinic Cell Carcinomas Display Similar Genomic Profiles, with Recurrent TP53 Mutations
Massively parallel sequencing yielded comparable coverage between microglandular adenosis (median 195 ×, range 47–903 ×) and acinic cell carcinoma (median 235 ×, range 100–661 ×) samples (Kolmogorov-Smirnov test, P>0.05). The two cases of pure microglandular adenosis displayed low levels of genetic instability (Figures 2 and 3a, Supplementary Table S2). Both cases were TP53 wild-type and harbored few copy number changes and few non-synonymous somatic mutations (MGA1, three mutations; MGA20, two mutations), which were all subclonal (Supplementary Figure S1, Supplementary Table S2). Nevertheless, a clonal synonymous mutation affecting SPTA1 was found in MGA1,9 suggesting that pure microglandular adenoses, although distinct from and not as genetically advanced as carcinoma-associated microglandular adenoses/ atypical microglandular adenoses, may constitute clonal neoplastic lesions. These results highlight the genetic heterogeneity of microglandular adenosis and expand on the observations made with comparative genomic hybridization,7, 8 given that a subset of microglandular adenoses, in particular those not associated with cancer, were found not only to display flat copy number profiles but also may lack clonal somatic mutations.

Non-synonymous somatic mutations detected by targeted capture massively parallel sequencing in microglandular adenoses and acinic cell carcinomas. Heatmap indicating the non-synonymous somatic mutations identified in the pure microglandular adenoses (n=2), carcinoma-associated microglandular adenoses/atypical microglandular adenoses (n=8), and acinic cell carcinomas (n=8) analyzed. Each column represents one sample; mutated genes are reported in rows. Mutation types are color-coded according to the legend. The presence of loss of heterozygosity of the wild-type allele of a mutated gene is represented by a diagonal bar.

Repertoire of copy number alterations identified in microglandular adenoses and acinic cell carcinomas. (a) Heatmap depicting the copy number alterations identified in the pure microglandular adenoses (n=2), carcinoma-associated microglandular adenoses/atypical microglandular adenoses (n=8), and acinic cell carcinomas (n=8) analyzed. Samples are represented on the y-axis, copy number alterations are represented along the x-axis according to their respective genomic location. Light red: copy number loss; white: neutral; light blue: copy number gain; dark blue: amplification. (b) Frequency plots of recurrent gains and losses in acinic cell carcinomas (top) and carcinoma-associated microglandular adenoses/atypical microglandular adenoses (middle). Significant differences (Fishers’ exact test, P<0.05) are plotted in the bottom panel. On the y-axis the proportion of samples in which gains (green bars) or losses (purple bars) were identified is plotted according to genomic location (x-axis). (c) Frequency plots of recurrent amplifications in acinic cell carcinomas (top) and carcinoma-associated microglandular adenoses/atypical microglandular adenoses (middle). Significant differences (Fishers’ exact test, P<0.05) are plotted in the bottom panel. On the y-axis the proportion of samples in which amplifications (green bars) were identified is plotted according to genomic location (x-axis).
Given that pure microglandular adenoses were genetically distinct from carcinoma-associated microglandular adenoses/atypical microglandular adenoses we excluded the two pure microglandular adenosis samples from further analyses, and compared eight carcinoma-associated microglandular adenoses/atypical microglandular adenoses (only microglandular adenosis/atypical microglandular adenosis components) with eight acinic cell carcinomas (only acinic cell components for those mixed acinic cell carcinomas). The overall non-synonymous somatic mutation rate was comparable between carcinoma-associated microglandular adenoses/atypical microglandular adenoses (median: 4.5, range 1–13) and acinic cell carcinomas (median: 4.0; range 1–7; Mann–Whitney U test, P>0.05), which also did not reveal any significant differences in single gene comparisons (Fisher’s exact tests, P>0.05; Figure 2, Supplementary Table S2). The mutational analysis (single-nucleotide variants and insertions and deletions) revealed significant inter-tumor heterogeneity within both groups, which, akin to triple-negative breast cancers, were characterized by several somatic mutations, but few recurrently mutated genes. TP53 was the only highly frequently mutated gene in microglandular adenoses/atypical microglandular adenoses (6/8, 75%) and acinic cell carcinomas (7/8, 87.5%). The majority of TP53 mutations were clonal (100% in microglandular adenoses/atypical microglandular adenoses and 75% in acinic cell carcinomas; Supplementary Figure S1) and in all cases where TP53 was affected by somatic mutations, loss of heterozygosity of the wild-type allele was detected (Figure 2), indicating complete inactivation of p53. TP53 mutations were either missense (microglandular adenosis/atypical microglandular adenosis 5/6; acinic cell carcinoma 5/7) or frame-shift, and all but one affecting the DNA-binding domain (Figure 4a, Supplementary Table S2). Recurrent alterations present in microglandular adenoses/atypical microglandular adenoses but not in acinic cell carcinomas included PTEN (2/8; M134I and H93R) and MED12 (2/8; D1204E and W439L) somatic mutations, whereas those restricted to acinic cell carcinomas included somatic mutations affecting ERBB4 (2/8; G6V and c.2203-1G-T). Finally, somatic mutations affecting BRCA1 (E1419* coupled with loss of heterozygosity), FGFR2 (S252W), ERBB3 (R667S), INPP4B (N223Y) and PIK3CA (E542Q) genes, among others, were observed in single cases of either group. It should be noted that the lack of significant differences between microglandular adenoses and acinic cell carcinomas may at least in part stem from the small sample size due to the rarity of these lesions.

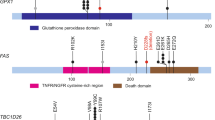
Comparison of somatic mutations detected by targeted capture massively parallel sequencing in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas cases with those in triple-negative breast cancers from The Cancer Genome Atlas. (a) Lollipop plots illustrating the prevalence and type of TP53 mutations in acinic cell carcinomas (top, n=8), carcinoma-associated microglandular adenoses/atypical microglandular adenoses (middle, n=8) and triple-negative breast cancers from The Cancer Genome Atlas (bottom, n=77). The structure of p53 protein is shown with a transactivation domain (green), a DNA-binding domain (black) and a tetramerization domain (blue). Each ‘lollipop’ represents a mutation that occurs at the amino acid location labeled along the x-axis. The height of each lollipop indicates the frequency of the mutation and the color of the lollipop represents the type of mutation. Mutation types are color-coded according to the legend. (b) Bar plots depicting the prevalence and type of mutations affecting other genes, detected in acinic cell carcinomas, carcinoma-associated microglandular adenoses/atypical microglandular adenoses and triple-negative breast cancers from The Cancer Genome Atlas. Each row represents a mutated gene; horizontal bars represent the prevalence of mutations affecting each gene in triple-negative breast cancers from The Cancer Genome Atlas (left) and carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas (right). Sample histologic types are color-coded according to the legend. Mutation types are texture-coded according to the legend. Owing to the high number of genes mutated at low frequency in triple-negative breast cancers, only genes affected by mutations in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas carcinomas, and genes recurrently mutated (n≥2) in triple-negative breast cancers are plotted. TCGA, The Cancer Genome Atlas.
A copy number alteration analysis revealed that all carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas displayed rather complex genomic profiles, with multiples regions of gains and losses and few focal high-level amplifications (Figure 3a). Recurrent copy number alterations in both groups included gains of 1q, 2q, 7p, and 8q and losses of 3p, 5q, 6q, 14q, 17p, and 17q. Notably, 16q whole-arm loss was absent; this lack of 16q losses is consistent with previously reported observations of the lack of 16q deletions in grade 3 triple-negative breast cancers.36 Amplifications of regions within 8q were present in both cohorts, including two microglandular adenoses/atypical microglandular adenoses (MGA7 and MGA8), where the amplicons encompassed FSBP, one of which extended to EPPK1 (MGA7), and one acinic cell carcinoma sample (ACC17), exhibiting an amplicon spanning MYC, SLA, and COL14A1. Consistent with the analysis of somatic mutations, only two focal regions were significantly differentially altered between microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas. 8q was significantly more frequently gained in microglandular adenoses/atypical microglandular adenoses, whereas 17q was significantly more frequently lost in acinic cell carcinomas (Fisher’s exact test P<0.05; Figure 3b). No region was significantly differentially amplified between the microglandular adenosis/atypical microglandular adenosis and acinic cell carcinoma groups (Fisher’s exact test P>0.05; Figure 3c).
As an exploratory, hypothesis generating analysis, we compared five invasive carcinomas arising in microglandular adenosis/atypical microglandular adenosis with four non-acinic cell carcinoma components of mixed acinic cell carcinomas. Their overall non-synonymous somatic mutation rates were similar (microglandular adenosis-associated carcinomas, median 5, range 3–10; non-acinic cell acinic cell carcinomas, median 4, range 3–8; Mann–Whitney U test, P>0.05). No significant differences were detected in terms of mutations (Fisher’s exact tests, P>0.05, Supplementary Figures S2 and S3) and only minimal differences in the repertoire of copy number alterations were detected between microglandular adenosis-associated invasive carcinomas and non-acinic cell components of acinic cell carcinomas (Supplementary Figure S4). 6q was significantly more frequently gained in non-acinic cell components of acinic cell carcinomas, while 8q was significantly more frequently gained in microglandular adenosis-associated invasive carcinomas, respectively (Supplementary Figure S4B), but no region was significantly differentially amplified or lost between these two subgroups (Supplementary Figure S4C). As another hypothesis generating analysis, we interrogated whether the acquisition of additional genetic alterations would be responsible for the progression of microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas as a group to invasive/non-acinic cell carcinomas. A comparison of the microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas components (n=16) versus the associated triple-negative breast cancer components (ie, microglandular adenosis-associated invasive carcinomas and non-acinic cell carcinoma components (n=9)) did not reveal significant differences in their overall non-synonymous somatic mutation rate (microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas, median 4, range 1–13; microglandular adenosis-associated invasive carcinomas/non-acinic cell acinic cell carcinomas, median 5, range 3–10; Mann–Whitney U test, P>0.05), or in mutations affecting single genes (Fisher’s exact tests, P>0.05). The copy number alteration analysis showed that focal regions on 1q, 17q, and 19q were significantly more frequently gained, but not amplified, in high-grade invasive/non-acinic cell carcinomas (Supplementary Figure S5, Fisher’s exact tests, P<0.05) than in microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas. Given the small sample size, additional and more comprehensive analyses of larger cohorts may be required to demonstrate differences between high-grade carcinomas arising in microglandular adenosis and acinic cell carcinomas, as well as the potential genetic underpinning of the histologic dedifferentiation and worse behavior of those high-grade triple-negative breast cancer components. Moreover, it should be noted that whilst clonal relatedness was previously demonstrated between all microglandular adenosis samples and co-existing invasive carcinomas included in this study,9 two non-acinic cell components were found not to be clonally related to the matched acinic cell carcinoma.12
Taken together, our data suggest that carcinoma-associated microglandular adenoses/atypical microglandular adenoses, acinic cell carcinomas and their co-existing invasive/non-acinic cell carcinoma components display a similar repertoire of somatic genetic alterations and, akin to conventional triple-negative breast cancers, are genetically heterogeneous, underpinned by complex patterns of copy number alterations and recurrent TP53 mutations. Our findings support the contention that microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas may constitute a spectrum of low-grade triple-negative breast cancers.
Carcinoma-Associated Microglandular Adenoses/Atypical Microglandular Adenoses and Acinic Cell Carcinomas Display Genomic Profiles Similar to those of Conventional Triple-Negative Breast Cancers
Given that carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas display important similarities in their repertoire of somatic genetic alterations and a triple-negative phenotype, we sought to compare their genomic profiles as a group (eight microglandular adenoses/atypical microglandular adenoses+eight acinic cell carcinomas) with those of 77 triple-negative breast cancers from The Cancer Genome Atlas. A comparison of the repertoire of somatic mutations did not reveal any significant difference between these two subgroups (Fisher’s exact tests, P>0.05, Figure 4). The TP53 gene was mutated in 81% of microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas and in 86% of The Cancer Genome Atlas triple-negative breast cancers, with no significant differences in the mutational spectrum (Figure 4a). Additional recurrent somatic mutations included those affecting PIK3CA and BRCA1, which were both mutated in 12.5% of microglandular adenosis/atypical microglandular adenosis/acinic cell carcinoma samples and in 10% and 5.5% of The Cancer Genome Atlas triple-negative breast cancers, respectively (Figure 4b). Copy number alteration analysis revealed both microglandular adenoses/ atypical microglandular adenoses and acinic cell carcinomas harbor recurrent 5q losses and 8q gains, which are typically observed in triple-negative breast cancers (Figure 5a). Few genomic regions differentially gained or lost between the two groups were identified (Fisher’s exact tests, P<0.05). Copy number alterations significantly more prevalent in microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas included gains of 2q, 7p, 15q, and 22p, whereas those significantly more frequent in The Cancer Genome Atlas triple-negative breast cancers included gains of 3q, 9p, 10p, 11q, and 13q, and losses of 4p, 10q, and 16q. The most significant differences mapped to 2q and 7p, which were both gained in >50% of microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas and in around 10% of The Cancer Genome Atlas triple-negative breast cancers (Figure 5a). Gains of 2q encompassed STAT1 and ERBB4 genes, whereas gains of 7p encompassed NEDL1 and ABCA13 genes. These changes were in the form of low-level gains and no region was significantly differentially amplified (Figure 5b). It should be noted that previous microarray-based comparative genomic hybridization analysis of an independent series of 12 pure or carcinoma-associated microglandular adenoses/atypical microglandular adenoses carried out by our group reported on the presence of recurrent gains of 2q and 7p in around 50% of cases,7 whereas in a study of 95 grade 3 invasive ductal carcinomas of no special type using the same microarray-based comparative genomic hybridization platform and similar bioinformatics methods, 2q and 7p were gained in around 20% of 25 triple-negative breast cancers.37 These previous results provide an independent validation of the significant differences in copy number profiles between microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas and triple-negative breast cancers from The Cancer Genome Atlas. It is plausible that activation of genes mapping to those regions play a role in the development of microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas.

Comparison of copy number alterations identified in carcinoma-associated microglandular adenosis/atypical microglandular adenosis and acinic cell carcinoma cases with those identified in triple-negative breast cancers from The Cancer Genome Atlas. (a) Frequency plots of recurrent gains and losses in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas (top, n=16) and triple-negative breast cancers from The Cancer Genome Atlas (middle, n=77). Significant differences (Fishers’ exact test P<0.05) are plotted in the bottom panel. On the y-axis the proportion of samples in which gains (green bars) or losses (purple bars) were identified is plotted according to genomic location (x-axis). (b) Frequency plots of recurrent amplifications in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas (top) and triple-negative breast cancers from The Cancer Genome Atlas (middle). Significant differences (Fishers’ exact test P<0.05) are plotted in the bottom panel. On the y-axis the proportion of samples in which amplifications (green bars) were identified is plotted according to genomic location (x-axis). TCGA, The Cancer Genome Atlas.
In contrast, a comparison between the mutation rates in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas, as a group, and ER-positive/HER2-negative (n=252) or HER2-positive (n=91) breast cancers from The Cancer Genome Atlas revealed statistically significant differences (Figure 6). Although PIK3CA is significantly less frequently mutated in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas than in ER-positive/HER2-negative tumors (12.5% versus 42%), the opposite was observed for mutations affecting the cancer genes TP53 (81% versus 22%), BRCA1 (12.5% versus 0.3%), INPP4B (12.5% versus 0.7%), ERBB4 (12.5% vsersu 0.3%), and FGFR2 (12.5% versus 0.7%, P<0.05, Fisher’s exact test). A similar comparison with HER2-positive breast cancers revealed only TP53 (81% versus 49%) and INPP4B (12.5% versus 0; P<0.05, Fisher’s exact test) to be significantly more frequently mutated in microglandular adenosis/atypical microglandular adenosis/acinic cell carcinoma samples. Less overt differences between our samples and HER2-positive tumors should be expected given that HER2-positive tumors include a mixture of ER-positive and ER-negative cancers, and can be stratified into two subgroups with distinct molecular characteristics according to ER status.14

Comparison of the frequencies of mutations affecting selected genes in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas, as a group, and in ER-positive/HER2-negative, HER2-positive and triple-negative breast cancers from The Cancer Genome Atlas (TCGA). ER, estrogen receptor.
Discussion
Here we provide a genetic basis for the substantial phenotypic overlap between microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas, in support of the notion that these two entities constitute a spectrum of low-grade triple-negative neoplasms. Carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas, in spite of their low-grade morphology, fully recapitulate the genomic features and genetic diversity of conventional high-grade triple-negative breast cancers, and show immunohistochemical serous acinar differentiation. Akin to triple-negative breast cancers,14 the only highly recurrently mutated gene in carcinoma-associated microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas is TP53. Furthermore, both entities displayed mutations affecting PI3K pathway genes (eg, PIK3CA, PTEN and INPP4B) and BRCA1, which are also recurrent in conventional triple-negative breast cancers.14
It is well accepted that breast cancer evolution follows two main pathways, which were initially stratified according to histologic grade.38, 39 The existence of a low-grade breast neoplasia family has been put forward;40 this family of lesions encompassed most well-established precursors of ER-positive breast cancers (flat epithelial atypia, atypical ductal hyperplasia and atypical lobular hyperplasia) and low-grade lobular and ductal in situ and invasive carcinomas.41 These lesions share a common immunophenotype (ER-positive, HER2-negative, high molecular weight cytokeratin-negative and low KI67 proliferation fraction) and genetic signature (highly recurrent PIK3CA mutations/1q gain/16q loss).38 We have later hypothesized that breast cancer evolution would be better stratified according to the expression of ER and ER-regulated genes,38 given that their expression defines two fundamentally distinct subgroups of breast cancer and that this genetic signature of low-grade ER-positive lesions is also observed at least in a subset of high-grade ER-positive breast cancers.14, 36
Although it is recognized that ER-positive breast cancers comprise a heterogeneous group of tumors and that progression from low- to high-grade ER-positive cancers occurs,36 ER-negative breast cancers have been considered a class of aggressive high-grade cancers. Our current findings showing that microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas are underpinned by similar genomic landscapes suggest the existence of a low-grade breast neoplasia family also in the ER-negative branch (Figure 7) that includes microglandular adenosis, atypical microglandular adenosis and acinic cell carcinoma. Based on their phenotypic and genetic similarities and given that pure microglandular adenoses do not always harbor copy number alterations7, 8 and seem to lack TP53 mutations9 and p53 overexpression,4, 5 microglandular adenosis may be a non-obligate direct precursor of both atypical microglandular adenosis and acinic cell carcinoma. It could be hypothesized that acquisition of TP53 mutation possibly represents a driver of progression to an atypical/malignant phenotype in the majority of cases, enabling the acquisition of a complex pattern of somatic mutations and copy number alterations, a hallmark feature of triple-negative breast cancers. In contrast, the early genetic events responsible for the development of microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas have yet to be defined. More comprehensive analyses (whole-genome, whole-exome and/or RNA sequencing) of larger cohorts of microglandular adenosis and acinic cell carcinoma samples may help ascertain whether microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas are underpinned by a single highly recurrent somatic mutation/fusion gene or represent a convergent phenotype caused by distinct genetic alterations.

Hypothetical evolution model of triple-negative breast neoplasms. Microglandular adenosis, atypical microglandular adenosis and acinic cell carcinoma likely constitute a low-grade triple-negative breast neoplasia family (middle pathway), characterized by recurrent TP53 mutations, 5q losses and 8q gains, and high levels of genetic instability, hallmark features of conventional high-grade triple-negative breast cancers (top pathway). Despite the lack of a myoepithelial cell layer, microglandular adenosis and atypical microglandular adenosis are not considered invasive lesions; therefore the progression from ductal carcinoma in situ to microglandular adenosis/atypical microglandular adenosis remains hypothetical as indicated by the dashed lines. Nevertheless, evidence favoring clonal relatedness between ductal carcinoma in situ and microglandular adenosis/atypical microglandular adenosis has been documented.7, 9 A second group of low-grade triple-negative neoplasms of the breast is underpinned by specific/pathognomonic genetic alterations, display low to intermediate levels of genetic instability and can be broadly categorized as salivary gland-like tumors of the breast, encompassing, most likely among others, secretory and adenoid cystic carcinomas. These special histologic types of triple-negative breast cancer are underpinned by ETV6-NTRK3 and MYB-NFIB fusion genes, respectively. *In their salivary gland counterparts, distinct genetic alterations but with likely similar functional effect have been described, such as MYBL1-NFIB fusion gene in adenoid cystic carcinomas54 and ETV6 rearrangements with an unknown partner (ETV6-X) in mammary-analog secretory carcinomas,55 which theoretically also occur in the breast lesions. ACC, acinic cell carcinoma; AMGA, atypical microglandular adenosis; DCIS, ductal carcinoma in situ; MGA, microglandular adenosis; TN, triple-negative; TNBC, triple-negative breast cancer. Solid lines, associations between lesions supported by molecular evidence; dashed line, hypothetical evolutionary associations.
It should be noted that another unrelated subset of triple-negative low-grade breast lesions exists, which can be broadly categorized as salivary gland-like tumors of the breast and encompasses histological entities underpinned by specific genetic alterations regardless of the anatomical site of origin, including adenoid cystic (MYB-NFIB) and secretory (ETV6-NTRK3) carcinomas (Figure 7), and possibly polymorphous adenocarcinoma and mucoepidermoid carcinoma, given that their salivary gland counterparts are underpinned by recurrent PRKD1 hotspot mutations42 and rearrangements of the MAML2 gene,43 respectively. Interestingly, the salivary gland counterpart of breast secretory carcinoma (aka, mammary analog secretory carcinoma) has only been recognized due to the identification of the specific fusion gene in lesions previously classified as unusual variants of salivary gland acinic cell carcinomas.44 In contrast to microglandular adenoses/atypical microglandular adenoses and breast acinic cell carcinomas, salivary gland-like tumors of the breast do not resemble conventional triple-negative breast cancers at the genetic level, harbor low levels of genetic instability with few copy number changes12, 45 and do not harbor highly recurrent TP53 mutations.12 Although those lesions may also, albeit less frequently, progress to high-grade triple-negative breast cancers, there is evidence that those high-grade carcinomas also differ genetically from conventional triple-negative breast cancers.45, 46 Previous observations based on histologic and immunohistochemical analyses have suggested a potential association between microglandular adenosis and adenoid cystic carcinomas.47 We cannot completely rule out that rare cases of salivary gland-like lesions of the breast might be clonaly related to co-existing microglandular adenosis and/or acinic cell carcinoma; however, molecular evidence to support this hypothesis has yet to be provided. Upon a detailed histologic review, none of the cases included in this study displayed histologic areas suggestive of adenoid cystic or secretory carcinoma worth to be tested for the presence of the respective specific fusion genes. Furthermore, our previous study demonstrating that acinic cell carcinomas of the breast and salivary glands are not related lesions48 corroborates the notion that breast acinic cell carcinomas should not be included in the low-grade salivary gland-like subgroup of triple-negative breast neoplasms. Finally, additional rare low-grade triple-negative breast cancers exist such as low-grade spindle fibromatosis-like and adenosquamous carcinomas, which may constitute yet a distinct subgroup of low-grade triple-negative breast cancers.49
Given that morphologic distinction between microglandular adenoses, in particular atypical microglandular adenoses, and pure acinic cell carcinomas can be challenging and that both entities harbor nearly identical molecular features to the best of our knowledge, one may question whether their differentiation would be of clinical relevance. Currently there are no optimal studies that provide a definitive answer to this question and whether microglandular adenoses/atypical microglandular adenoses not associated with carcinoma and pure low-grade acinic cell carcinomas behave differently. Although the last WHO classification states that some histological, immunohistochemical, and ultrastructural differences favor the distinction of microglandular adenosis/atypical microglandular adenosis and acinic cell carcinoma,11 our data showing diffuse expression of lysozyme in microglandular adenosis/atypical microglandular adenosis cells calls into question the practice of differentiating these two entities on the basis of the expression of serous differentiation markers. Moreover, akin to microglandular adenoses and atypical microglandular adenoses,5 acinic cell carcinomas may also display circumferential expression of antibodies targeting basement membrane such as laminin.13 Therefore, to establish a diagnosis of pure acinic cell carcinoma of microglandular growth pattern, it might be prudent to use criteria similar to those used to diagnose invasive carcinoma in microglandular adenoses/atypical microglandular adenoses, such as the presence of stromal desmoplasia and/or confluent glands.
Although microglandular adenoses/atypical microglandular adenoses can be considered lesions with uncertain malignant potential,50 acinic cell carcinoma is an indolent form of triple-negative breast cancer and may not mandate systemic treatment unless associated with a high-grade component or diagnosed at advanced stage. Until observational studies with larger cohorts provide evidence-based treatment recommendations, we would argue that the therapeutic implications of both diagnoses, in particular for local control, can be similar.7, 13 Conlon et al.13 have recently described two breast acinic cell carcinomas and suggested that microglandular adenosis-like tubular areas in acinic cell carcinomas should be interpreted as part of the carcinomatous process and therefore re-excision performed if that process extends to the initial margins. We suggest that similar principles should be applied to carcinoma-associated microglandular adenoses/atypical microglandular adenoses and margins free of microglandular adenosis/atypical microglandular adenosis should be achieved.
Therapy of pure microglandular adenoses, which vary from microscopic to extensive foci, is less straightforward due to the lack of consistent data on their rate of progression to carcinoma and the rate of biopsy underestimation. Recently, Bois et al.51 analyzed a series of 13 538 benign breast biopsies and found pure microglandular adenosis in 21 (0.2%) cases. Subsequent invasive carcinoma was diagnosed in only one patient that displayed concurrent atypical ductal hyperplasia, suggesting that the rate of progression may be extremely low, in a way akin to what has been described for flat epithelial atypia, the first morphologically identifiable precursor of the low-grade ER-positive breast neoplasms.38, 52, 53 The contention, however, that a benign lesion has potential to progress into an aggressive malignant tumor has led some authors to suggest that modification of the current management approach of microglandular adenosis may be necessary.6, 7, 8, 9, 10 Given that microglandular adenosis may coexist with and may be genetically as advanced as acinic cell carcinomas and conventional triple-negative breast cancers, we concur with the notion that a diagnosis of pure microglandular adenosis in needle-biopsies should trigger a surgical excision to rule out the presence of an associated malignancy. Completeness of excision should be interpreted in the appropriate context with consideration of the presence, degree and extent of atypia, and the presence of associated lesions.
Our study has several limitations. First, we cannot completely rule out that the few significant differences observed between microglandular adenoses and acinic cell carcinomas and between these two entities as a group and conventional triple-negative breast cancers are due to the small number of samples analyzed. The rarity of these lesions renders the accrual of larger series for massively parallel sequencing analysis challenging. Importantly, however, we were able to demonstrate statistically significant differences between microglandular adenosis/acinic cell carcinomas and ER-positive/HER2-negative or HER2-positive breast cancers. Second, our targeted sequencing panel is limited to genes previously known to be altered in breast cancer or to be associated with DNA repair. It is possible that somatic genetic alterations affecting gene(s) not included in the targeted sequencing panel employed here and/or epigenetic alterations may constitute founder events in microglandular adenosis/atypical microglandular adenosis and/or acinic cell carcinoma and possibly associated with their indolent behavior. Further studies based on whole genome, whole exome and/or RNA sequencing analysis of microglandular adenoses/atypical microglandular adenoses and acinic cell carcinomas are warranted. Finally, due to the multi-institutional nature of our cohorts and the association of most cases with high-grade triple-negative breast cancers, we do not have follow-up information, nor sufficient power to interrogate differences between pure microglandular adenoses and pure acinic cell carcinomas in terms of clinical behavior. In addition, the prognosis of high-grade triple-negative breast cancers arising in microglandular adenosis or acinic cell carcinoma remains to be defined and may differ from that of conventional triple-negative breast cancers.
Despite these limitations, our study is the first to provide molecular evidence in favor of the contention that microglandular adenosis and acinic cell carcinoma are related lesions, harboring similar genomic profiles and recurrent TP53 mutations. In addition, their profiles do not differ substantially from common forms of triple-negative breast cancers, except for focal and low-level copy number alterations, including gains of 2q and 7p, which were preferentially found in microglandular adenoses/atypical microglandular adenoses/acinic cell carcinomas. More comprehensive studies are required to confirm our findings and to define whether activation of gene(s) mapping to these regions or alternative genetic alterations may promote their development. Taken together our results provide evidence to put forward the existence of a low-grade triple-negative breast neoplasia family constituted by microglandular adenosis, atypical microglandular adenosis and acinic cell carcinoma, which represent low-grade forms of triple-negative disease with no/minimal metastatic potential that may progress to high-grade triple-negative breast cancers.
References
Shin SJ, Gobbi H et al. Microglandular adenosis, atypical microglandular adenosis and microglandular adenosis with carcinoma. In: Lakhani SR, Ellis IO, Schnitt SJ, (eds). WHO Classification of Tumours of the Breast. IARC Press: Lyon, France, 2012, pp 113–114.
Rosen PP . Microglandular adenosis. A benign lesion simulating invasive mammary carcinoma. Am J Surg Pathol 1983;7:137–144.
Rosenblum MK, Purrazzella R, Rosen PP . Is microglandular adenosis a precancerous disease? A study of carcinoma arising therein. Am J Surg Pathol 1986;10:237–245.
Khalifeh IM, Albarracin C, Diaz LK et al. Clinical, histopathologic, and immunohistochemical features of microglandular adenosis and transition into in situ and invasive carcinoma. Am J Surg Pathol 2008;32:544–552.
Koenig C, Dadmanesh F, Bratthauer GL et al. Carcinoma Arising in Microglandular Adenosis: an Immunohistochemical Analysis of 20 Intraepithelial and Invasive Neoplasms. Int J Surg Pathol 2000;8:303–315.
Geyer FC, Kushner YB, Lambros MB et al. Microglandular adenosis or microglandular adenoma? A molecular genetic analysis of a case associated with atypia and invasive carcinoma. Histopathology 2009;55:732–743.
Geyer FC, Lacroix-Triki M, Colombo PE et al. Molecular evidence in support of the neoplastic and precursor nature of microglandular adenosis. Histopathology 2012;60:E115–E130.
Shin SJ, Simpson PT, Da Silva L et al. Molecular evidence for progression of microglandular adenosis (MGA) to invasive carcinoma. Am J Surg Pathol 2009;33:496–504.
Guerini-Rocco E, Piscuoglio S, Ng CK et al. Microglandular adenosis associated with triple-negative breast cancer is a neoplastic lesion of triple-negative phenotype harbouring TP53 somatic mutations. J Pathol 2016;238:677–688.
Tsang JY, Tse GM . Microglandular adenosis: a prime suspect in triple-negative breast cancer development. J Pathol 2016;239:129–132.
Eusebi V, Ichihara S, Vincent-Salomon V et al. Exceptionally rare types and variants. In: Lakhani SR, Ellis IO, Schnitt SJ et al, (eds). WHO Classification of Tumours of the Breast. IARC Press: Lyon, France, 2012, pp 71–76.
Guerini-Rocco E, Hodi Z, Piscuoglio S et al. The repertoire of somatic genetic alterations of acinic cell carcinomas of the breast: an exploratory, hypothesis-generating study. J Pathol 2015;237:166–178.
Conlon N, Sadri N, Corben AD et al. Acinic cell carcinoma of breast: morphologic and immunohistochemical review of a rare breast cancer subtype. Hum Pathol 2016;51:16–24.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70.
Kahn R, Holtveg H, Nissen F et al. Are acinic cell carcinoma and microglandular carcinoma of the breast related lesions? Histopathology 2003;42:195–196.
Peintinger F, Leibl S, Reitsamer R et al. Primary acinic cell carcinoma of the breast: a case report with long-term follow-up and review of the literature. Histopathology 2004;45:645–648.
Ng CK, Schultheis AM, Bidard FC et al. Breast cancer genomics from microarrays to massively parallel sequencing: paradigms and new insights. J Natl Cancer Inst 2015;107: pii: djv015.
Subhawong AP, Subhawong T, Nassar H et al. Most basal-like breast carcinomas demonstrate the same Rb-/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am J Surg Pathol 2009;33:163–175.
Osako T, Takeuchi K, Horii R et al. Secretory carcinoma of the breast and its histopathological mimics: value of markers for differential diagnosis. Histopathology 2013;63:509–519.
Piscuoglio S, Ng CK, Murray MP et al. The Genomic Landscape of Male Breast Cancers. Clin Cancer Res 2016;22:4045–4056.
Cibulskis K, Lawrence MS, Carter SL et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219.
Saunders CT, Wong WS, Swamy S et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–1817.
Koboldt DC, Zhang Q, Larson DE et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–576.
Robinson JT, Thorvaldsdottir H, Winckler W et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26.
De Mattos-Arruda L, Weigelt B, Cortes J et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol 2014;25:1729–1735.
Schwarz JM, Rödelsperger C, Schuelke M et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575–576.
Carter H, Chen S, Isik L et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res 2009;69:6660–6667.
Martelotto LG, Ng CK, De Filippo MR et al. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol 2014;15:484.
Kandoth C, McLellan MD, Vandin F et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–339.
Futreal PA, Coin L, Marshall M et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–183.
Lawrence MS, Stojanov P, Mermel CH et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495–501.
Shen R, Seshan VE . FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016; doi:10.1093/nar/gkw520 (e-pub ahead of print).
Curtis C, Shah SP, Chin SF et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–352.
Carter SL, Cibulskis K, Helman E et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30:413–421.
Landau DA, Carter SL, Stojanov P et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013;152:714–726.
Natrajan R, Lambros MB, Geyer FC et al. Loss of 16q in high grade breast cancer is associated with estrogen receptor status: evidence for progression in tumors with a luminal phenotype? Genes Chromosome Cancer 2009;48:351–365.
Natrajan R, Lambros MB, Rodriguez-Pinilla SM et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin Cancer Res 2009;15:2711–2722.
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M et al. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology 2010;57:171–192.
Roylance R, Gorman P, Hanby A et al. Allelic imbalance analysis of chromosome 16q shows that grade I and grade III invasive ductal breast cancers follow different genetic pathways. J Pathol 2002;196:32–36.
Abdel-Fatah TM, Powe DG, Hodi Z et al. Morphologic and molecular evolutionary pathways of low nuclear grade invasive breast cancers and their putative precursor lesions: further evidence to support the concept of low nuclear grade breast neoplasia family. Am J Surg Pathol 2008;32:513–523.
Rakha E . The low nuclear grade breast neoplasia family. Diagn Histopathol 2012;18:124–132.
Weinreb I, Piscuoglio S, Martelotto LG et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet 2014;46:1166–1169.
O'Neill ID . t(11;19) translocation and CRTC1-MAML2 fusion oncogene in mucoepidermoid carcinoma. Oral Oncol 2009;45:2–9.
Skalova A, Vanecek T, Sima R et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol 2010;34:599–608.
Del Castillo M, Chibon F, Arnould L et al. Secretory Breast Carcinoma: a histopathologic and genomic spectrum characterized by a joint specific ETV6-NTRK3 gene fusion. Am J Surg Pathol 2015;39:1458–1467.
Fusco N, Geyer FC, De Filippo MR et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod Pathol 2016; doi:10.1038/modpathol.2016.134 (e-pub ahead of print).
Acs G, Simpson JF, Bleiweiss IJ et al. Microglandular adenosis with transition into adenoid cystic carcinoma of the breast. Am J Surg Pathol 2003;27:1052–1060.
Piscuoglio S, Hodi Z, Katabi N et al. Are acinic cell carcinomas of the breast and salivary glands distinct diseases? Histopathology 2015;67:529–537.
Geyer FC, Lambros MB, Natrajan R et al. Genomic and immunohistochemical analysis of adenosquamous carcinoma of the breast. Mod Pathol 2010;23:951–960.
Rakha EA, Badve S, Eusebi V et al. Breast lesions of uncertain malignant nature and limited metastatic potential: proposals to improve their recognition and clinical management. Histopathology 2016;68:45–56.
Bois CM, Al-Hilli Z, Visscher DW et al. Microglandular adenosis and risk of breast cancer: a Mayo benign breast disease cohort study. Mod Pathol 2016;29:32A.
Boulos FI, Dupont WD, Simpson JF et al. Histologic associations and long-term cancer risk in columnar cell lesions of the breast: a retrospective cohort and a nested case-control study. Cancer 2008;113:2415–2421.
Aroner SA, Collins LC, Schnitt SJ et al. Columnar cell lesions and subsequent breast cancer risk: a nested case-control study. Breast Cancer Res 2010;12:R61.
Gonda TJ, Ramsay RG . Adenoid cystic carcinoma can be driven by MYB or MYBL1 rearrangements: new insights into MYB and tumor biology. Cancer Discov 2016;6:125–127.
Skalova A, Vanecek T, Simpson RH et al. Mammary analogue secretory carcinoma of salivary glands: molecular analysis of 25 ETV6 gene rearranged tumors with lack of detection of classical ETV6-NTRK3 fusion transcript by standard RT-PCR: report of 4 cases harboring ETV6-X gene fusion. Am J Surg Pathol 2016;40:3–13.
Acknowledgements
CM is funded in part by AIRC (MFAG13310). Research reported in this paper was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Geyer, F., Berman, S., Marchiò, C. et al. Genetic analysis of microglandular adenosis and acinic cell carcinomas of the breast provides evidence for the existence of a low-grade triple-negative breast neoplasia family. Mod Pathol 30, 69–84 (2017). https://doi.org/10.1038/modpathol.2016.161
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.161
This article is cited by
-
Molecular pathology in breast disease: diagnostic, prognostic, and therapeutic tools
Virchows Archiv (2024)
-
Problematic breast tumors reassessed in light of novel molecular data
Modern Pathology (2021)
-
Morphologic and immunohistochemical features of carcinoma involving microglandular adenosis of the breast following neoadjuvant chemotherapy
Modern Pathology (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}