
Abstract
African and African-American (AA) women have higher incidence of triple-negative breast cancers (TNBC) with high histological grade and aggressive clinical behavior, but the reasons are not fully understood. We recently found that the oncogenic protein EZH2 is overexpressed in Ghanaian breast cancer patients, with 16% of the tumors expressing cytoplasmic EZH2. Understanding the molecular underpinnings of these aggressive tumors may lead to the identification of potential targetable oncogenic drivers. We characterized the copy number variations of 11 Ghanaian breast tumor patients by targeted multiplexed PCR-based DNA next-generation sequencing (NGS) over 130 cancer-relevant genes. While the DNA quality was not optimal for mutation analysis, 90% of the tumors had frequent recurrent copy number alterations (CNAs) of 17 genes: SDHC, RECQL4, TFE3, BCL11A, BCL2L1, PDGFRA, DEK, SMUG1, AKT3, SMARCA4, VHL, KLF6, CCNE1, G6PD, FGF3, ABL1, and CCND1, with the top oncogenic functions being mitotic G1–G1/S-phase regulation, gene transcription, apoptosis, and PI3K/AKT pathway. The most common recurrent high-level CNAs were gains of RECQL4 and SDHC, in 50% and 60% of cases, respectively. Network analyses revealed a significant predicted interaction among 12 of the 17 (70.6%) genes with high-level CNAs (p = 5.7E−07), which was highly correlated with EZH2 expression (r = 0.4–0.75). By immunohistochemistry, RECQL4 and SDHC proteins were upregulated in 53 of 86 (61.6%) and 48 of 86 (56%) of Ghanaian invasive carcinoma tissue samples. In conclusion, our data show that invasive carcinomas from Ghana exhibit recurrent CNAs in 17 genes, with functions in oncogenic pathways, including PI3K/AKT and G1–G1/S regulation, which may have implications for the biology and treatment of invasive carcinomas in African and AA women.
Similar content being viewed by others


Introduction
Breast cancer is the second most incident cancer worldwide, accounting for over 2 million new cases in 2018 [1]. African women have a lower lifetime incidence of breast cancer than women from North America or Europe; however, age-standardized mortality is poorer, particularly for women from Western Africa [1]. This likely reflects a combination of environmental and systematic factors, as well as underlying differences in tumor biology. Women from Sub-Saharan Africa tend to have earlier breast cancer presentation [2, 3]. Histologically, invasive breast carcinomas from this population are frequently basal-like and are negative for estrogen receptor (ER), progesterone receptor (PR), and HER2/neu overexpression and amplification (i.e., triple-negative) [4]. Similarly, African-American (AA) women, who are thought to share African ancestry, have significantly poorer outcomes than white patients after accounting for age, stage, and socioeconomic status, and are more likely to have breast cancer earlier and ER negative status [5,6,7,8]. The underlying molecular alterations for the aggressive breast cancer phenotypes of women of African ancestry are not fully understood.
Basal-like breast cancers are a heterogeneous group of tumors comprising 15–20% of all breast cancers. They are more common in premenopausal, and in African and AA women [9]. We previously reported on the incidence of triple-negative breast cancers (TNBC) in Ghanaian women (53.2%), which more closely resembled AA women (30%) than white American women (15.5%), suggesting that African ancestry may correlate with a higher likelihood of TNBC [10]. Similar results have been found in other cohorts of women from Senegal and Nigeria [6]. The higher proportion of TNBC in invasive breast tumors from African and AA patients is associated with ALDH1 and AR expression, suggesting novel subtypes of TNBC in these populations [11], but the genetic landscapes need investigation.
Enhancer of Zeste Homolog 2 (EZH2), an epigenetic regulator responsible for transcriptional repression, is overexpressed in multiple tumor types [12,13,14,15,16,17]. In breast cancer, overexpression of EZH2 is significantly associated with negative ER and PR status and distant metastasis [12, 17, 18]. We previously showed that EZH2 is significantly associated with high-grade and basal-like tumors in a cohort of 169 breast tissues from Ghanaian women [16]. We detected EZH2 expression in the cytoplasm of 16% of Ghanaian invasive carcinomas where it was significantly associated with TNBC status [16]. While cytoplasmic EZH2 has been shown to increase breast cancer invasion and metastasis, the relevance of cytoplasmic EZH2 to the aggressive behavior of breast cancer in African women has not been studied.
Despite advances in our understanding of the genomic landscape of breast and other cancers, the genetic alterations in invasive carcinomas from African women are far from understood. In this study, we define a set of somatic copy number alterations (CNAs) in a cohort of histologically well-characterized Ghanaian invasive breast carcinomas, and investigate associations with patterns of EZH2 subcellular localization.
Materials and methods
Human tissue samples and TMA development
Tumor samples were collected from 100 women with breast cancer who underwent surgery at Komfo Anoyke Teaching Hospital (KATH) in Kumasi, Ghana, between 2006 and 2011. Clinicopathological features of these tumors, ER, PR, HER2, and EZH2 immunostaining, were previously reported [16]. Formalin-fixed, paraffin-embedded tissues were stained with H&E and reviewed independently by the study pathologists (MR and CK), and tumors were arrayed in a high-density tissue microarray (TMA) with triplicate samples (n = 255 TMA samples) following established protocols [12]. We selected 20 cases for next-generation sequencing (NGS) analyses based on the following criteria: (i) sufficient tissue on the block, (ii) representative samples with EZH2 expression in the nucleus, cytoplasm, or no expression, and (iii) representative samples with different histology, tumor grade (2–3), hormonal receptor and HER2/neu status.
Targeted NGS
Targeted NGS of breast tumor tissue was performed with IRB approval. For each specimen, 4–10 × 10-µm formalin-fixed, paraffin-embedded sections of 20 breast tissue samples were cut from a single block and macrodissected with a scalpel under guidance of an H&E-stained slide to enrich for tumor content. We isolated DNA using the Qiagen Allprep FFPE DNA/RNA kit (Qiagen, Valencia, CA). DNA was quantified using the Qubit 2.0 fluorometer (Life Technologies, Foster City, CA).
Targeted, multiplexed PCR-based NGS was performed on each tumor. For samples with DNA concentration of >40 ng, we used the Ion Ampliseq Comprehensive Cancer Panel (CCP), which targets 1,688,650 bases from 15,992 amplicons representing 409 cancer genes (Thermo Fisher Scientific). For samples with <40 ng of DNA, we employed the Oncomine Comprehensive Panel (OCP) (Thermo Fisher Scientific), which is compatible with 20 ng of formalin-fixed paraffin-embedded isolated DNA and benchtop Ion Torrent sequencers [19]. Barcoded libraries were generated from 40 ng of DNA per sample using the CCP, or 20 ng of DNA per sample using the OCP, and the Ion Ampliseq Library Kit 2.0 (Life Technologies, Foster City, CA) according to the manufacturer’s instructions with barcode incorporation. Sequencing of template libraries was then performed on an Ion Proton sequencer with Ion PI chips (Thermo Fisher Scientific) using the Ion PI Hi-Q Sequencing 200 Kit v3 (Life Technologies, Foster City, CA) according to the manufacturer’s instructions. Data analysis was performed as described previously in Torrent Suite (version 5.0.4) and in-house previously validated bioinformatics pipelines [19, 20].
Copy number analysis
Amplicon-level read counts were determined using the coverage analysis plugin. Briefly, normalized GC content corrected read counts per amplicon for each sample were divided by those from a composite normal male DNA sample (composed of multiple formalin-fixed, paraffin-embedded and frozen tissues, individual and pooled samples) to generate amplicon-level copy number ratios, and weighted gene-level copy number estimates were determined as described previously [20]. Genes with a log2 copy number ratio estimate of <−1 or >0.80 were considered to have high-level loss (deletion) or gain, respectively.
Network and bioinformatics analyses
For enrichment analyses, we performed gene ontology (GO) overrepresentation tests (GO annotations: biological process, molecular function, cellular compartment, and protein domain) in PANTHER (v14.1) and STRING (v11.0) database for confirming enrichment results with topological features from protein/gene interaction networks. A p value of < 0.05 was significant. Correlation between EZH2 and the 17 genes with CNAs was analyzed using Pearson correlation coefficient (r).
Immunohistochemistry for RECQL4 and SDHC
Immunohistochemistry was performed on the TMA containing 100 invasive carcinomas from Ghanaian women. Four-micron-thick sections of the TMA were prepared for staining with H&E and immunohistochemistry using a standard biotin–avidin complex technique, anti-RECQL4 (Abcam, ab188125, rabbit polyclonal, 1:50) and anti-SDHC (Abcam, ab155999 rabbit monoclonal, 1:2500) antibodies. Nuclear expression of RECQL4 and cytoplasmic expression of SDHC was interpreted independently by two pathologists (MR and CK). For each case the staining was graded as negative (score = 1, no staining); weak (score 2, <25% of cells staining, any intensity); moderate (score = 3, 25–75% of cells staining, any intensity); and strong (score = 4, >75% of cells staining, any intensity) following previous studies [12, 21]. High RECQL4 or SDHC expression was defined as scores 3 and 4, and low expression was defined as scores 1 and 2.
Results
NGS identifies frequent CNAs in invasive breast carcinomas from Ghanaian women
Targeted NGS was performed on 20 formalin-fixed, paraffin-embedded breast tissue samples from Ghana identified with sufficient tissue, 9 were excluded due to poor DNA quality. The final cohort consisted of 11 samples comprising 10 invasive carcinomas and 1 fibroadenoma control. Representative micrographs are shown in Fig. 1a–f.

Low- (a, c, e) and high-power (b, d, f) hematoxylin and eosin-stained (H&E) sections from three invasive carcinomas, Sample BR13 (a, b) is an invasive ductal carcinoma of intermediate grade with mucinous differentiation, Sample BR19 (c, d) is an invasive ductal carcinoma of grade 3, and Sample BR22 (e, f) is metaplastic carcinoma with squamous differentiation and histological grade 3.
All patients were female, ranging from 38 to 64 years old (mean, 48 years). Of the invasive carcinomas, seven were invasive ductal of intermediate and high histological grade, and three were metaplastic carcinomas with squamous differentiation. EZH2 expression was high in the nucleus or cytoplasm (three and six cases), and was negative in one tumor. Eight invasive carcinomas were TNBC including the metaplastic carcinomas, one was HER2/neu positive, and one was luminal B (positive for ER, PR, and HER2/neu). The fibroadenoma sample was negative for expression of EZH2. Details on the clinical and histologic characteristics of this cohort are listed in Table 1.
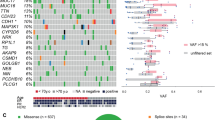
Using CCP and OPC NGS platforms according to the DNA concentration of each tumor, we successfully analyzed CNAs in ten invasive carcinomas and one fibroadenoma tissue sample. The quality of the Ghanaian samples was not adequate for accurate sequencing studies. Copy number analysis of NGS data yielded a total of 17 high-level CNAs. Prioritized high-level CNAs for each case are shown in an integrative heatmap (Fig. 2).

Shown are 17 high-level copy number alterations identified and color coded as indicated in the figure, using CCP and OCP NGS platforms. In all, 90% of the invasive carcinomas show CNAs, largely gains, with the most frequent being RECQL4 and SDHC. Note that CNAs in PDGFRA and DEK are identified in tumors with nuclear EZH2, while CNAs in BCL2L1, AKT3, SMARCA4, CCND1, FGF3, and ABL1 are detected in tumors with cytoplasmic EZH2.
Copy number analysis of NGS data demonstrated recurrent CNAs, including most frequent gain of chromosomes 1q (SDHC, AKT3) 8q (RECQL4), and X (TFE3, G6PD) and loss of chromosome 9q (ABL1). Nine (90%) invasive carcinomas demonstrated CNAs of at least one of these genes: SDHC, RECQL4, TFE3, BCL11A, BCL2L1, PDGFRA, DEK, SMUG1, AKT3, SMARCA4, VHL, KLF6, CCNE1, G6PD, FGF3, and CCND1. One case showed loss of ABL1. It is interesting to note that we identified gains in RECQL4, PDGFRA, AKT3, and SMARCA4 in the fibroadenoma, suggesting that these alterations occur in early neoplastic lesions. Together, these data identify CNAs in invasive carcinomas from Ghanaian women with biological and translational implications.
Bioinformatics reveals a predicted interaction network among genes with CNAs and EZH2 expression
Out of 17 genes, 9 (53%) with CNAs were detected in invasive carcinomas with nuclear or cytoplasmic EZH2 expression. Gains in PDGFRA oncogene and loss of DEK tumor suppressor were identified in invasive carcinomas with only nuclear EZH2, and gains in BCL2L1, AKT3, SMARCA4, and CCND1 as well as gains and losses of FGF3, and loss of ABL1 were detected in invasive carcinomas with only cytoplasmic EZH2 expression (Fig. 2).
We next tested the hypotheses that the genes harboring frequent recurring CNAs may be functionally related, and that they may associate with EZH2 expression. Enrichment analysis in GO annotation and STRING databases revealed a significantly enriched predicted interaction network (p value = 5.76E−07) among 12 of the 17 genes with CNAs (70.6%), as well as a correlation between these genes and EZH2 (Pearson correlation coefficient r = 0.4–0.75) (Fig. 3a). Analyses using GO annotation for the top 10 altered pathways (REACTOME) showed significant representation of mitotic G1–G1/s phases (ABL1, AKT3, CCND1, and CCNE1), transcription pathway (ABL1, AKT3, CCND1, CCNE1, DEK, G6PD, and SMARCA4), PI3K/AKT signaling in cancer (AKT3, FGF3, and PDGFRA), and RMTs methylate histone arginines (CCND1 and SMARCA4) (Fig. 3b and Table 2). Collectively, these data suggest a relationship between the 17 genes with CNAs and EZH2 expression and localization, which warrants further investigation. Our results highlight major oncogenic pathways deregulated in Ghanaian invasive carcinomas that can be explored for therapeutic application.

a Network graph shows enrichment (p value = 5.7E−07) of topological features of the 17 genes with CNAs. Enrichment analysis in STRING database (v11.0) predicts interactions between EZH2 and 12 of the 17 genes with CNAs (r = 0.4–0.75). b. Enrichment analyses with biological functions of top ten altered biological pathways (REACTOME). Legend (bars: −log (p value).
RECQL4 and SDHC show frequent copy number gains and protein overexpression in Ghanaian invasive carcinomas
Genes demonstrating the highest frequency of copy number gains in our cohort were SDHC (1q23.3) and RECQL4 (8q24.3), neither of which has been previously considered in the context of breast carcinoma in African patients. SDHC, which encodes Succinate Dehydrogenase Complex Subunit C, was amplified in 6 of 10 (60%) invasive carcinomas, and RECQ4L, encoding a DNA helicase and functions in homologous recombination-mediated double-stranded break repair, was amplified in 5 of 10 (50%) carcinoma cases sequenced (Figs. 2 and 4).

Copy number profiles for three Ghanaian invasive carcinomas with high-level CNAs highlighting gains at SDHC (1q23.3) and RECQL4 (8q24.3). Log2 copy number ratios per amplicon are plotted, with each individual amplicon represented by a single dot.
To explore whether CNAs in these genes are associated with protein overexpression, we next tested RECQL4 and SDHC protein expression in 100 tissue samples of Ghanaian invasive carcinomas arranged in TMAs. Of the 86 tumors with sufficient tissue to evaluate in the TMA, positive nuclear RECQL4 staining was detected in 53/86 (61.6%) and positive SDHC cytoplasmic expression in 48/86 (56%) tumors (Fig. 5a-c). Further supporting our data, analysis of the TCGA breast tissue data sets using UALCAN showed that RECQL4 and SDHC mRNA expression is significantly upregulated in invasive carcinomas compared with normal breast tissue samples (Supplementary Fig. 1A). When analyzed according to patient race, RECQL4 mRNA expression is significantly higher in invasive carcinomas in AA women compared to Caucasians and Asians, and to normal tissues, while SDHC transcript was significantly higher in Caucasians and Asians compared to AAs (Supplementary Fig. 1B). Analysis of TCGA breast cancer database suggests that high mRNA levels of RECQL4 and SDHC may be associated with worse overall survival compared to low mRNA levels (Supplementary Fig. 1C).

Representative images of invasive carcinomas with negative and positive nuclear RECQL4 (a) and cytoplasmic SDHC (b) expression, from the TMA containing 86 invasive carcinomas from Ghana. c Quantification of the percentage of invasive carcinomas negative and positive for each protein.
Discussion
While African women have a lower lifetime incidence in breast cancer, age-standardized mortality is poorer [1]. AA women, like women from Ghana, have a higher incidence of TNBC and basal-like tumors than white American women, suggesting that African ancestry may be associated with differences in tumor biology [10]. Our laboratory reported that invasive carcinomas from Ghana have a significantly higher frequency of high histological grade, triple-negative status, and EZH2 overexpression [16]. However, the genetic alterations that underlie the aggressive biological behavior of invasive carcinomas in African women have not been fully elucidated, in part due to the limited availability of tissue samples from these patients. Here, we used NGS to discover high frequency CNAs in invasive carcinomas from Ghana, which may influence their biological behavior and advance our understanding of these tumors.
Our studies revealed the presence of recurrent CNAs in 17 genes in 90% of invasive carcinomas from Ghana studied, including gains in SDHC, RECQL4, TFE3, BCL11A, BCL2L1, PDGFRA, AKT3, SMARCA4, VHL, KLF6, CCNE1, G6PD, FGF3, and CCND1, gains and losses in DEK, SMUG1, and FGF3, and loss of ABL1. Functionally, these genes belong to critical signaling pathways in breast tumorigenesis, including PI3K-Akt, transcriptional, and cell cycle regulatory pathways, which have not been previously considered in African breast cancer. These data pave the way to investigating the contribution of these signaling pathways to the aggressive clinical behavior of breast cancer in this population, and to design studies testing the potential utility of inhibiting these pathways to halt breast cancer progression.
Emerging data suggest that in addition to transcriptional repression, EZH2 exerts oncogenic functions in a subset of TNBCs by interacting with cytoplasmic proteins that regulate cell migration and invasion [18]. Our lab has reported that EZH2 is overexpressed in the nucleus in 42% and in the cytoplasm in 16% of Ghanaian invasive carcinomas, where it is associated significantly with TNBC status [16]. We have recently shown that activation of p38 mitogen activated kinase leads to EZH2 phosphorylation at threonine 367 with resulting accumulation of EZH2 protein in the cytoplasm and binding with vinculin and other cytoskeletal proteins [18]. Of the 17 genes with CNAs identified in this study, 6 (35%) are associated with cytoplasmic (gains of BCL2L1, AKT3, SMARCA4, and CCND1 as well as gains and losses of FGF3, and loss of ABL1) and 2 (12%) with nuclear EZH2 expression (gains in PDGFRA oncogene and loss of DEK tumor suppressor). Network analyses demonstrated a robustpredicted interaction between EZH2 and these genes, especially strong with DEK, SMARCA4, CCNE1, ABL1, and CCND1. These data are intriguing as there are several reports of EZH2-mediated regulation of these genes. For example, EZH2 has been reported to directly repress DEK in fibroblasts [22], and EZH2 and PDGFRA were found inversely associated in Merkel cell carcinomas [23], EZH2 inhibition was found to selectively kill SMARCA4 deficient cells in small cell carcinoma of the ovary [24], and EZH2 was shown to regulate cell cycle progression and the levels of cyclins in various cancer types, including our studies showing that EZH2 regulates CCND1 [25,26,27,28].
Our study shows that the most frequently altered genes in Ghanaian breast cancer patients are RECQL4 and SDHC, harboring copy number gains in 50% and 60% of invasive carcinomas in our cases. These findings were validated in a histopathologically well-characterized cohort of invasive carcinomas treated at the KATH in Kumasi, Ghana, where RECQL4 and SDHC proteins were overexpressed in 61.6% and 56% of 86 tumors with sufficient tissue for evaluation.
RECQL4 is mapped to 8q24, a gene desert flanked by MYC and PVT1. Amplification of this region is frequently associated with susceptibility to multiple tumor types, including breast and prostate [29, 30]. The mechanism of this susceptibility has been thought to occur through the amplification of MYC [31] or ncRNA PVT1 [32]. In our study, we observed MYC amplification in 2/11 samples compared to RECQL4 amplification in 6/11 samples, suggesting that in this cohort, gains of RECQL4 may occur independently of MYC. Several studies have demonstrated through loss-of-function in vitro studies that RECQL4 contributes to breast cancer proliferation and chemoprotection [33, 34]. Succinate dehydrogenase protein C (SDHC) is part of a family of metabolic enzymes that function in the citric acid cycle and electron transport chains. Germline inactivating mutations in SDHC result in accumulation of succinate and are associated with paragangliomas, renal cell carcinomas, and gastrointestinal stromal tumors [35]. While RECQL4 and SDHC have been studied in the context of breast cancer, they have not been previously considered in African populations.
The current study evidences the difficulties encountered in studying invasive breast tumors from western Sub-Saharan populations. While we began our study with 20 FFPE blocks of Ghanaian breast tumors, 9 were excluded due to low quality DNA and 3 additional had low DNA content requiring the use of a smaller sequencing panel. Despite these limitations, our team has generated a cohort of breast tissue samples from Ghana with adequate material for molecular studies. We have identified novel high-level CNAs in 17 genes with functions in tumorigenic pathways and associations to the oncoprotein EZH2. We validated the frequent overexpression of RECQL4 and SDHC tumors in this patient population. Collectively, these data provide the basis for further sequencing and clinical studies to better understand the pathobiology and offer therapeutic opportunities for breast cancer in African and AA women.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Fregene A, Newman LA. Breast cancer in Sub-Saharan Africa: how does it relate to breast cancer in African-American women? Cancer. 2005;103:1540–50.
Alluri P, Newman LA. Basal-like and triple-negative breast cancers: searching for positives among many negatives. Surg Oncol Clin N Am. 2014;23:567–77.
Stark A, Kleer CG, Martin I, Awuah B, Nsiah-Asare A, Takyi V, et al. African ancestry and higher prevalence of triple-negative breast cancer: findings from an international study. Cancer. 2010;116:4926–32.
Shavers VL, Harlan LC, Stevens JL. Racial/ethnic variation in clinical presentation, treatment, and survival among breast cancer patients under age 35. Cancer. 2003;97:134–47.
Huo D, Ikpatt F, Khramtsov A, Dangou JM, Nanda R, Dignam J, et al. Population differences in breast cancer: survey in indigenous African women reveals over-representation of triple-negative breast cancer. J Clin Oncol. 2009;27:4515–21.
Huo D, Hu H, Rhie SK, Gamazon ER, Cherniack AD, Liu J, et al. Comparison of breast cancer molecular features and survival by African and European Ancestry in The Cancer Genome Atlas. JAMA Oncol. 2017;3:1654–62.
Newman LA. Breast cancer in African-American women. Oncologist. 2005;10:1–14.
Toft DJ, Cryns VL. Minireview: basal-like breast cancer: from molecular profiles to targeted therapies. Mol Endocrinol. 2011;25:199–211.
Jiagge E, Oppong JK, Bensenhaver J, Aitpillah F, Gyan K, Kyei I, et al. Breast cancer and African ancestry: lessons learned at the 10-year Anniversary of the Ghana-Michigan Research Partnership and International Breast Registry. J Glob Oncol. 2016;2:302–10.
Proctor E, Kidwell KM, Jiagge E, Bensenhaver J, Awuah B, Gyan K, et al. Characterizing breast cancer in a population with increased prevalence of triple-negative breast cancer: androgen receptor and ALDH1 expression in Ghanaian women. Ann Surg Oncol. 2015;22:3831–5.
Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11.
Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–73.
Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, et al. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360–70.
Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19:86–100.
Pang J, Toy KA, Griffith KA, Awuah B, Quayson S, Newman LA, et al. Invasive breast carcinomas in Ghana: high frequency of high grade, basal-like histology and high EZH2 expression. Breast Cancer Res Treat. 2012;135:59–66.
Ding M, Zhang H, Li Z, Wang C, Chen J, Shi L, et al. The polycomb group protein enhancer of zeste 2 is a novel therapeutic target for cervical cancer. Clin Exp Pharm Physiol. 2015;42:458–64.
Anwar T, Arellano Garcia C, Ropa J, Chen YC, Kim HS, Yoon E, et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat Commun. 2018;9:2801.
Hovelson DH, McDaniel AS, Cani AK, Johnson B, Rhodes K, Williams PD, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015;17:385–99.
Cani AK, Hovelson DH, McDaniel AS, Sadis S, Haller MJ, Yadati V, et al. Next-Gen sequencing exposes frequent MED12 mutations and actionable therapeutic targets in phyllodes tumors. Mol Cancer Res. 2015;13:613–9.
Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9.
Iannetti A, Ledoux AC, Tudhope SJ, Sellier H, Zhao B, Mowla S, et al. Regulation of p53 and Rb links the alternative NF-kappaB pathway to EZH2 expression and cell senescence. PLoS Genet. 2014;10:e1004642.
Veija T, Koljonen V, Bohling T, Kero M, Knuutila S, Sarhadi VK. Aberrant expression of ALK and EZH2 in Merkel cell carcinoma. BMC Cancer. 2017;17:236.
Chan-Penebre E, Armstrong K, Drew A, Grassian AR, Feldman I, Knutson SK, et al. Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: vitro and in vivo preclinical models Mol Cancer Ther. 2017;16:850–60.
Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, et al. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene. 2009;28:843–53.
Li X, Gonzalez ME, Toy K, Filzen T, Merajver SD, Kleer CG. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol. 2009;175:1246–54.
Cao J, Pontes KC, Heijkants RC, Brouwer NJ, Groenewoud A, Jordanova ES, et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. J Pathol. 2018;245:433–44.
Cao W, Feng Z, Cui Z, Zhang C, Sun Z, Mao L, et al. Up-regulation of enhancer of zeste homolog 2 is associated positively with cyclin D1 overexpression and poor clinical outcome in head and neck squamous cell carcinoma. Cancer. 2012;118:2858–71.
Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21.
Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7.
Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V, et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer. 2006;6:245.
Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13:5745–55.
Fang H, Nie L, Chi Z, Liu J, Guo D, Lu X. et al. RecQL4 helicase amplification is involved in human breast tumorigenesis. PLoS ONE. 2013;8:e69600.
Arora A, Agarwal D, Abdel-Fatah TM, Lu H, Croteau DL, Moseley P, et al. RECQL4 helicase has oncogenic potential in sporadic breast cancers. J Pathol. 2016;238:495–501.
Aldera AP, Govender D. Gene of the month: SDH. J Clin Pathol. 2018;71:95–7.
Acknowledgements
This work was supported by the NCI of the NIH under award numbers R01CA125577 and R01CA107469 (to C. Kleer), F30CA19084 (to T. Anwar), the University of Michigan Rogel Cancer Center support grant P30CA046592, and the Department of Defense award W81XWH-19-1-0093 (to C. Kleer). We thank members of the Kleer lab for useful discussions during the execution of the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
ST has had a prior sponsored research agreement with Thermo Fisher Scientific. He is the co-founder of, prior consultant to, equity holder in, and current employee of Strata Oncology. All other authors have no relevant conflicts of interest to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This study was presented as a platform presentation at the 2019 108th USCAP Annual Meeting.
Supplementary information
Rights and permissions
About this article

Cite this article
Anwar, T., Rufail, M.L., Djomehri, S.I. et al. Next-generation sequencing identifies recurrent copy number variations in invasive breast carcinomas from Ghana. Mod Pathol 33, 1537–1545 (2020). https://doi.org/10.1038/s41379-020-0515-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0515-2
This article is cited by
-
Characterization and clinical relevance of PDGFRA pathway copy number variation gains across human cancers
Molecular Genetics and Genomics (2022)
