
Abstract
At mucosal surfaces, phagocytes such as macrophages coexist with microbial communities; highly controlled regulation of these interactions is essential for immune homeostasis. Pattern-recognition receptors (PRRs) are critical in recognizing and responding to microbial products, and they are subject to negative regulation through various mechanisms, including downregulation of PRR-activating components or induction of inhibitors. Insights into these regulatory mechanisms have been gained through human genetic disease–association studies, in vivo mouse studies utilizing disease models or targeted gene perturbations, and in vitro and ex vivo human cellular studies examining phagocytic cell functions. Although mouse models provide an important approach to study macrophage regulation, human and mouse macrophages exhibit differences, which must be considered when extrapolating mouse findings to human physiology. This review discusses inhibitory regulation of PRR-induced macrophage functions and the consequences of dysregulation of these functions and highlights mechanisms that have a role in intestinal macrophages and in human macrophage studies.
Similar content being viewed by others
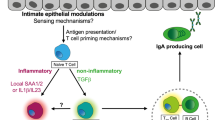

Introduction
Following microbial exposure, the peripheral immune system must mount responses to limit infection and clear microbes. Mucosal surfaces, such as the intestine, continually interact with microbes1 and therefore must balance the mechanisms defending against pathogens or excessive entry of resident microbiota with mechanisms maintaining tolerance to resident microbiota. To maintain this balance, anti-microbial responses undergo tight regulation. Uncontrolled inflammation can lead to the development of autoimmune and inflammatory diseases. However, overactive inhibitory mechanisms can increase susceptibility to infections and decrease microbial clearance, which can lead to persistent inflammation. Initial microbial recognition and responses occur through innate cells, particularly phagocytes. Consistently, depleting or altering the proportion and phenotype of these cells modulates severity of multiple autoimmune and inflammatory diseases, including at mucosal surfaces.2, 3, 4, 5 Phagocytes are comprised of myeloid-derived cells such as macrophages, dendritic cells (DCs) and neutrophils;6 this review will focus on regulation in macrophages, although the regulation in DCs can be similar7 and will occasionally be highlighted.
Macrophages regulate microbes at multiple levels, including through immune mediator secretion, microbial killing, pyroptosis, adaptive immune instruction, and wound healing.6 Macrophages recognize and respond to microbial components through pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs), nucleotide-binding-domain containing receptors (NLRs), retinoic-acid-inducible-gene I-like receptors, and C-type lectins.8 Distinct PRR can share specific signaling pathways but can also signal through diverse intermediates. For example, TLR2, TLR5, TLR7, and TLR9 utilize the myeloid differentiation primary response gene 88 (MyD88)-dependent pathway, which activates nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK).9 Interleukin-1 receptor (IL-1R) and IL-18R also utilize this pathway. By contrast, TLR3 signals through MyD88-independent or Toll/interleukin1-domain-containing adaptor-inducing interferon-β (TRIF) pathways.9 TLR4 utilizes both MyD88-dependent and -independent pathways.9 Although multiple PRR share pathways, the outcomes can vary dramatically.9 Contributions to this variability include different adaptor molecule combinations, subcellular PRR localization and strength of signaling; however, many mechanisms accounting for the distinct outcomes are unclear.9
PRR engagement in macrophages induces pro-inflammatory and anti-inflammatory cytokines; the balance between these cytokines influences outcomes.9 Pro- and anti-inflammatory cytokines can be distinctly regulated, as can pro-inflammatory cytokines themselves (e.g., distinct inflammasome-mediated regulation of IL-1 and IL-18 secretion). Importantly, PRR-induced cytokine regulation varies over time. For example, initial pro-inflammatory cytokine induction can be followed by anti-inflammatory cytokines and other inhibitory mechanisms, thereby contributing to inflammation resolution. Notably, altered expression of various PRR-pathway inhibitors is observed in tissue and sera in many human inflammatory diseases. However, the role of these alterations is often unknown and may be either secondary to direct dysregulation or to compensatory regulation.
Human polymorphisms in regions containing PRR-pathway genes and their regulators in macrophages have been associated with multiple autoimmune/inflammatory diseases, including inflammatory bowel disease (IBD). Polymorphisms in regions involving PRR or PRR-initiated signaling pathways (e.g., NOD2, IRF5, NFKB1, RELA, REL, RIPK2, CARD9, PTPN22), paracrine/autocrine cytokine pathways modifying PRR signaling (e.g., IL-23R, IL-12, IL-10, IL-18RAP/IL1R1, IFNGR2/IFNAR1, JAK2, STAT3, TYK2), and autophagy pathways (e.g., ATG16L1 and IRGM) are associated with IBD.10 Mouse studies provide essential mechanistic insight into how pathways in phagocytic cells mediate dysregulation in vivo; conditional ablation of pertinent genes in myeloid cells further elucidates select myeloid cell functions.7 While sharing similarities, mouse and human phagocytic cells also exhibit differences in microbial responses, cytokine induction and differentiation patterns.6, 11, 12, 13 These differences must be considered when extrapolating mouse results to human studies.12 Consequently, parallel primary human macrophage studies are necessary. This review will focus on the inhibitory mechanisms regulating phagocytic cell functions and will particularly emphasize human studies and relevance to human inflammatory diseases. Where applicable, we will highlight the relevance of such mechanisms to mucosal surfaces, particularly the intestine.
Monocyte Migration and Tissue-Dependent Differentiation
Monocytes are derived from a granulocyte monocyte-forming unit that differentiates in the bone marrow through cytokines, including monocyte-stimulating factor and granulocyte/monocyte-stimulating factor;13 the transcription factor PU.1 regulates this differentiation.13 Differentiated monocytes exit the bone marrow, enter the blood, and then migrate into tissues. In humans, circulating blood monocytes undergo apoptosis after 3–4 days,14 and must differentiate to prolong survival.15 Under homeostasis, monocytes enter tissues and become macrophages where they acquire functions integral to and characteristic of the tissues in which they reside.13 Macrophages are larger, more effective phagocytes, and survive longer than monocytes, living for up to several months.14
PRR-Induced Macrophage Functions Must be Tightly Controlled
Initial PRR-induced responses are critical in controlling microbial insults;1 however, these responses must be tightly regulated to prevent excessive cytokine secretion leading to systemic inflammatory response syndromes, including “endotoxin shock” or “sepsis,” which can result in tissue injury and death. Upon chronic microbial stimulation, PRR-induced cytokines, chemokines, and activation markers are downregulated.16, 17, 18, 19, 20 This process, termed “endotoxin tolerance”, has been most commonly described with chronic TLR4 stimulation.19 Mucosal macrophages encounter ongoing microbial exposure, and may be particularly subject to this type of regulation; multiple mechanisms contribute to reducing responsiveness to microbial products. Although some mechanisms limit acute PRR-mediated outcomes, these and other inhibitory mechanisms can be enhanced or induced following chronic microbial product stimulation. These mechanisms include downregulation of expression and/or function of PRR or critical intermediates in PRR-initiated pathways, modulation of strength of signaling, regulation by microRNAs, epigenetic regulation of gene promoters, as well as regulation by the inflammasome and autophagy (Figure 1 and Table 1). Below, we will emphasize select inhibitory mechanisms and highlight where appropriate their relevance to homeostasis at mucosal surfaces, including the intestine.

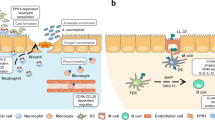
Mechanisms downregulating pattern-recognition receptor (PRR)-mediated macrophage responses. Signaling through PRR (e.g., Toll-like receptor 4 (TLR4)) activates distinct inflammatory pathways. The myeloid differentiation primary response gene 88 (MyD88)-dependent pathway signals through IRAK-1, nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK) pathways, while the MyD88-independent TRIF pathway signals through interferon regulatory factors (IRFs). Multiple mechanisms limit excessive macrophage responses induced by these pathways. They include: (1) downregulation of receptors and signaling molecules (e.g., PRRs, IRAK-1); (2) induction of decoy receptors (e.g., SIGIRR); (3) induction of intracellular or surface molecules that inhibit MyD88-dependent or -independent pathways; (4) induction of secretory molecules, including anti-inflammatory mediators (interleukin (IL)-10, transforming growth factor (TGF)-β, IL-1Ra), which suppress inflammation through multiple mechanisms; (5) signaling through alternative subunits or altering strength of signaling of molecules; (6) targeting of inflammatory RNAs by microRNAs thereby decreasing protein expression; (7) epigenetic regulation, including chromatin modification leading to inhibition of pro-inflammatory genes and/or upregulation of inhibitory genes; and (8) autophagy, which limits autocrine IL-1 secretion and therefore IL-1 contributions to secretion of additional pro-inflammatory cytokines. Inflammatory intermediates are indicated in green and inhibitory molecules/mechanisms in red. IFN, interferon; PI3K, phosphatidylinositol 3-kinase; TNF, tumor necrosis factor.
Regulating expression of PRR signaling pathway genes
PRR-initiated macrophage responses can be inhibited by downregulating the expression and/or function of PRR complexes or PRR-initiated signaling pathways (Figure 1 and Table 1). Although these mechanisms may decrease excessive inflammation and sepsis, they may also adversely affect bacterial clearance. PRR expression downregulation or receptor complex affinity alterations for microbial ligands during endotoxin tolerance have been observed in some, but not all, human and mouse macrophage studies.21, 22, 23 However, overexpressing TLR4 or TLR adaptor molecules fails to reverse downregulated cytokine secretion in certain situations,24, 25, 26 indicating that redundant endotoxin tolerance-inducing mechanisms can compensate for dysregulated modulation of receptor expression. Consistent with the requirement for TLR downregulation to limit intestinal PRR responses, epithelial cells and colitis-associated tumors from IBD patients and lamina propria macrophages from ulcerative colitis (UC) patients show increased TLR4 expression.27, 28 This may increase inflammation, as mice overexpressing TLR4 in epithelial cells are more susceptible to dextran sodium sulfate (DSS)-colitis and colitis-associated cancer.29
Downregulation or limited activation of PRR signaling intermediates, such as interleukin-1 receptor-associated kinase (IRAK)-1 and NF-κB (Figures 1 and 2 and Tables 1 and 2), further attenuates PRR-initiated responses. IRAK-1 participates in MyD88-dependent9, 30 and some MyD88-independent pathways.30, 31 IRAK-1 degradation following PRR stimulation limits subsequent inflammation in human monocytic cells.20, 31 Furthermore, in non-monocytic cells, such as mouse enterocytes, failure to downregulate IRAK-1 upon postnatal PRR ligand exposure may contribute to necrotizing enterocolitis.32 Consistent with IRAK-1 signaling downregulation preventing excessive inflammation, mice deficient in IRAK-4, a kinase that activates IRAK-1, are protected against endotoxin shock but are more susceptible to bacterial infections.33 IRAK-1 polymorphisms resulting in increased expression and/or kinase activity are associated with more severe sepsis.34, 35 By contrast, human IRAK-4 loss-of-expression mutations increase invasive pneumococcal disease risk.36 Downstream NF-κB signaling is critical for PRR-induced responses and its modulation changes PRR-initiated outcomes. For example, decreased phosphorylation and nuclear translocation of NF-κB subunits downregulates TLR4-mediated signaling in human intestinal macrophages.26 Although NF-κB activation is generally associated with inflammation, its outcomes are more complex; for example, NF-κB directly contributes to anti-microbial responses. Moreover, NF-κB has various regulatory roles, which vary in different intestinal cell subsets, and different NF-κB subunits have distinct functional roles. For example, while p50/p65 NF-κB heterodimers generally induce inflammatory pathways and these individual subunits are essential for proper anti-microbial responses,37, 38 p50/p50 homodimers inhibit inflammatory responses,39, 40 and contribute to endotoxin tolerance in mouse macrophages.41, 42 Similarly, mice deficient in IKKβ (Ikappa B kinase beta) in myeloid cells are more susceptible to endotoxin shock.43 Moreover, mice deficient in p50 and heterozygous for p65 develop spontaneous typhlocolitis.44 However, p65 knock down attenuates experimental colitis,45 and increased PRR-induced NF-κB activation in epithelial cells exacerbates colitis but only if accompanied with MAPK activation and tumor necrosis factor (TNF)-α production.46 Another complexity is that p50 is processed from the NFKB1 gene product p105,39 which exhibits additional regulatory functions, as p105-deficient mice expressing p50 still show increased lung and liver inflammation.47 Importantly, NFKB1 polymorphisms are associated with UC10 and necrotizing enterocolitis,48 and polymorphisms in the NF-κB subunits REL and RELA are associated with IBD;10 the functional consequences of these polymorphisms are unclear. In IBD patients, increased p65 expression and NF-κB-binding activity is seen in intestinal macrophages and epithelial cells.49, 50 Therefore, regulating the expression and function of PRR-signaling pathway intermediates in macrophages occurs on multiple levels. As both loss- and gain-of-function of these molecules can lead to inflammation, fine tuning their regulation is crucial for proper immune homeostasis.

Inhibitory molecules operate by numerous mechanisms to limit pattern-recognition receptor (PRR)-induced macrophage functions. Intracellular inhibitors regulate inflammatory responses on multiple levels by interfering with PRR-initiated signaling. Selected mechanisms of action of intracellular inhibitors are also summarized in Table 2. Inflammatory intermediates are indicated in green and inhibitory molecules in red. ABIN, A20 binding inhibitor of NF-κB; ERK, extracellular signal–regulated kinase; GAS, growth-arrest specific; IFNR, interferon receptor; IRF, interferon regulatory factor; ITAM, immunoreceptor tyrosine activation motif; JAK, Janus tyrosine kinase; MKP, mitogen-activated protein kinase phosphatase; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor κB; NLR, nucleotide oligomerization domain-like receptor; PI3K, phosphatidylinositol 3-kinase; SHIP, SH2-containing inositol 5′-phosphatase; SOCS, suppressor of cytokine signaling; TLR, Toll-like receptor; TRAF, TNF receptor–associated factor.
Inhibitory molecules regulate PRR signaling by various mechanisms
PRR-induced inflammation can be downregulated through inhibitors targeting PRR-initiated pathways (Figure 2 and Table 2). Some inhibitors are constitutively expressed and inhibit basal cytokine expression and/or initial PRR signaling modulation, whereas others are upregulated after PRR stimulation. Furthermore, inflammation can be regulated by several inhibitory waves following PRR stimulation.51 Inhibitors are often increased in inflamed tissues, as the immune system attempts to control the inflammation. Below, we will discuss examples of inhibition of PRR-initiated responses by surface receptor and intracellular inhibitory molecules. A summary of these and other inhibitors is shown in Figure 2, Tables 1 and 2.
Inhibitory surface receptors or secreted decoy receptors regulate PRR-induced inflammation. For example, initial studies found that ST2 sequesters MyD88 and Mal to prevent TLR4-dependent NF-κB signaling, cytokine induction, and endotoxin tolerance in vivo in mice,52, 53 as well as in human monocytes.54 Subsequently, membrane-bound ST2 (ST2L) was identified as a receptor for IL-33,55 and controversy ensued as to the composite effects of IL-33/ST2L interactions in inhibiting or activating cytokine-inducing pathways. These interactions may differentially affect inflammation in distinct cell types and tissues, including in the intestine.56, 57 In mice, IL-33 binds to ST2 on Th2 (T helper type 2) cells,57 and this interaction promotes Th2-mediated colitis.56 ST2 also exists as a soluble isoform, which is a decoy receptor that binds to IL-33 and inhibits its signaling,58 adding further complexity to the ST2-mediated regulation of inflammatory responses. Polymorphisms in a region containing ST2 (IL1RL1) are associated with Crohn’s disease (CD).59 In UC patients, inflamed mucosa and sera express increased soluble ST2, whereas ST2L expression on the surface of epithelial cells is downregulated, adding further complexity to elucidating the ultimate role of ST2 in IBD.56
IRAK-1 inhibition regulates not only PRR-, but also IL-1R- and IL-18R-induced pathways.9 Basal expression of the IRAK-1-activation inhibitor, IRAK-M, in mouse and human macrophages controls initial PRR-mediated inflammation.31, 60, 61 Chronic infection or PRR stimulation further upregulates IRAK-M in macrophages,31, 60, 62, 63 which, in turn, downregulates cytokine induction following PRR restimulation.31, 60, 62 This partially occurs by preventing IRAK-1 and IRAK-4 dissociation from MyD88, thereby abrogating further IRAK-1-dependent signaling.60 IRAK-M deficiency increases severity of endotoxin shock60 and of mucosal injury models in mice, including in the lung64 and colon.65 Loss-of-function polymorphisms have been associated with inflammatory/autoimmune diseases such as asthma,66 and dysregulated IRAK-M levels are seen in various diseases, including asthma,67 necrotizing enterocolitis,68 tuberculosis,69 and cystic fibrosis.62
TLR3 and TLR4 activate the MyD88-independent TRIF pathway, which induces Type I interferons (IFNs) and IFN-γ. Autocrine interferon-mediated inflammation is, in turn, regulated by the suppressor of cytokine signaling (SOCS) proteins, in particular SOCS-1, which primarily inhibits interferon-initiated JAK/STAT (Janus tyrosine kinase/signal transducer and activator of transcription factor) pathways.70, 71, 72, 73, 74 SOCS-1-deficient mice show early death due to multi-organ inflammation,73 and decreased survival to sublethal TLR4 stimulation.72 Initial papers described SOCS-1 as downregulating TLR4-induced NF-κB;72, 75 however, subsequent studies found the SOCS-1-deficient mouse phenotype was mostly attributable to excessive type I IFN inflammatory effects, rather than defects in direct TLR4 signaling inhibition by SOCS-1.76, 77 Mutations decreasing SOCS-1 expression are associated with immune dysregulation, including increased serum Immunoglobulin E78 and asthma.79 SOCS-1 upregulation can be mediated by the three TAM receptors expressed primarily in myeloid cells: Tyro3, Axl, and Mer.80, 81 Axl also upregulates the transcriptional repressor Twist1 in human macrophages.82 Mer-deficient mice are more susceptible to endotoxin shock.83 Deficiency in all the three TAM receptors and the subsequent lack of SOCS-1 induction results in uncontrolled macrophage-mediated pro-inflammatory cytokine secretion and autoimmunity.81 Besides inhibiting PRR-mediated inflammation,80 TAM receptors regulate DC chemotaxis84 and apoptotic cell phagocytosis and clearance by macrophages.85, 86, 87 Defects in TAM receptor-induced phagocytosis contribute to autoimmunity in TAM-deficient mice.85 Consistently, apoptotic cell ingestion is associated with anti-inflammatory cytokine production and tolerance.88 Increased levels of Protein S, a TAM receptor ligand, are seen in inflammatory diseases such as SLE (systemic lupus erythematosus) and UC;89, 90 the role of this elevation in the disease process is unclear.
An important inhibitor targeting multiple pathways, A20, was initially implicated in terminating TNF-induced NF-κB activation in mice91 through ubiquitinating a TNF signaling intermediate receptor-interacting protein (RIP) and targeting it for degradation.92 Subsequent studies demonstrated that A20 also inhibits PRR-initiated pathways such as the TLR4 pathway through ubiquitinating TRAF6 (TNF receptor–associated factor 6),93 the TLR3 pathway by preventing IRF3 (interferon regulatory factor 3) dimerization,94 and the nucleotide-binding oligomerization domain-containing protein 2 (NOD2) pathway by ubiquitinating and degrading the NOD2 downstream effector RIP2.95 A20-deficient mice die prematurely due to severe multi-organ inflammation.91 Moreover, selective A20 deficiency in DCs induces spontaneous colitis.96 Polymorphisms in the A20 region, including loss-of function mutations, are associated with multiple autoimmune/inflammatory diseases, including CD,97 SLE,98, 99 and rheumatoid arthritis (RA).100, 101 IRF4 inhibits PRR pathways in mouse and human monocytic cells by preventing RIP2102 and MyD88 signaling,103 and decreasing JNK and NF-κB activation.104 Consistently, IRF4−/− mice demonstrate more severe endotoxin shock.103 Furthermore, IRF4 upregulation upon chronic NOD2 stimulation contributes to protection from experimentally induced colitis,102 and IRF4 expression is increased in the mucosa of IBD patients.105
SH2-containing inositol 5'-phosphatase (SHIP-1) inhibits MyD88-independent pathways by dephosphorylating and inhibiting tank binding kinase 1 (TBK1), thereby modulating IFNβ induction.106 SHIP-1 also limits phosphatidylinositol 3-kinase (PI3K) signaling.107 SHIP-1-deficient mice are hyper-responsive to lipopolysaccharide (LPS) stimulation and defective in endotoxin tolerance.107 However, over time SHIP-1 deficiency promotes anti-inflammatory M2 macrophage polarization, possibly to counteract the inflammatory phenotype.108 SHIP-1−/− mice exhibit ileitis109 and increased macrophage infiltration into bone marrow and spleen, which ultimately decreases survival.110 Increased SHIP-1 expression is observed in the intestinal mucosa of IBD patients;111 it is unclear if this increase reflects a compensatory mechanism to counteract the inflammation.
FCγ receptors and β2-integrins containing ITAM domains downregulate multiple PRR pathways by inducing PRR signaling inhibitors, including SOCS-1, A20, and IL-10 in primary human macrophages.112 Consistently, ITAM receptor induction protects from inflammation, including experimental colitis.113, 114 Which specific ITAM-containing receptors mediate inhibitory effects and whether ITAM domain-containing peptides can be used therapeutically in inflammatory diseases has yet to be defined.
Secreted inhibitory mediators downregulate PRR-induced pro-inflammatory pathways in an autocrine and/or paracrine fashion
PRR stimulation results in secretion of autocrine/paracrine factors that can feed back to inhibit PRR pathways directly or indirectly, thereby suppressing inflammatory outcomes. For example, PRR-induced transforming growth factor (TGF)-β can inhibit pro-inflammatory cytokines by degrading their transcripts and suppressing their translation.115 TGF-β also inhibits NF-κB signaling,26 downregulates CD40116 and induces SHIP-1.107 IL-10 also regulates inflammation through multiple mechanisms. IL-10 decreases transcript and protein expression of numerous genes, including pro-inflammatory cytokines,115, 117, 118, 119 and leads to degradation of the MyD88-dependent signaling intermediates IRAK-1 and IRAK-4.120 IL-10 also upregulates inhibitors such as soluble TNFR, IL-1Ra and SOCS-3121 and upregulates STAT3 and PI3K pathways,122 which can inhibit inflammatory pathways.123, 124, 125, 126, 127, 128, 129, 130 Moreover, IL-10 inhibits additional myeloid cell functions, including phagocytosis and antigen presentation.123 Autocrine IL-10 and TGF-β signaling frequently combine to effectively suppress inflammation, including in human macrophages and at mucosal sites.18, 126, 131 Mouse studies have long confirmed the importance of these secretory mediators in homeostasis at mucosal surfaces: IL-10 and TGF-β-deficient mice develop spontaneous colitis.132 Depleting macrophages in IL-10-deficient mice attenuates the colitis,133 demonstrating that dysregulated macrophage function contributes to disease. Besides downregulating inflammation in macrophages proper, IL-10 production by gut macrophages suppresses inflammation by modulating other cell subsets, such as intestinal FoxP3+ T cells.134 Mice with deletion or loss-of-function of the TGF-β receptor in myeloid cells show impaired DSS-colitis resolution,135 indicating that besides producing TGF-β myeloid cells must also respond to TGF-β to limit intestinal inflammation. Consistently, TGF-β is required for downregulating pro-inflammatory cytokines in human lamina propria macrophages.136 Importantly, human polymorphisms in regions including IL10, IL10R, and SMAD3 are associated with autoimmune/inflammatory diseases, including IBD.10, 137, 138 An additional PRR-induced secretory mediator, IL-1Ra, inhibits IL-1R signaling.139 As autocrine/paracrine IL-1 dramatically enhances overall cytokine secretion by human macrophages, IL-1Ra ultimately downregulates multiple cytokines (Figure 1).140 Consistently, IL-1Ra deficiency or blockade worsens experimental colitis141 and increases endotoxin susceptibility in mice,142 and loss-of-function IL-1Ra polymorphisms are associated with autoimmune diseases143 (Table 1).
Emerging evidence shows that in specific circumstances, cytokines with pro-inflammatory roles such as IL-1β,126 interferons,82, 144 and TNF-α,61, 145, 146 can, in fact, also downregulate PRR- and FCγR-mediated inflammation in human macrophages. Mechanisms mediating these anti-inflammatory responses can include induction of certain inhibitory proteins and inhibitory signaling pathways (e.g., GSK3 (glycogen synthase kinase-3) signaling).61
Distinct isoforms and strength of signaling modulate PRR-induced macrophage outcomes
Targeting of MAPK and PI3K signaling pathways is being studied in therapeutic trials.147, 148 As modulating the quality and/or quantity of these pathways determines whether they activate or inhibit PRR-initiated pathways (Figure 1), an improved understanding of their signaling would more accurately predict therapeutic outcomes.
Extracellular signal–regulated kinase (ERK) can inhibit LPS-induced pro-inflammatory cytokines in mouse macrophages.149, 150 However, this inhibition has not generally been observed in human studies.151, 152 More recent studies in human macrophages identified that PRR-induced ERK indeed inhibits pro-inflammatory cytokines but that this inhibition is masked by autocrine IL-1, which then increases the strength of MAPK signaling and modifies the downstream outcomes.153 Differential susceptibility to lowering ERK signaling can be observed between different pro-inflammatory cytokines as well, such that IL-1β secretion decreases upon ERK inhibition more readily than TNF-α in LPS-stimulated mouse macrophages.154 Therefore, the strength of MAPK signaling can dramatically influence whether pro-inflammatory cytokine secretion is inhibited or enhanced in macrophages.
PI3K can both downregulate124, 125, 126, 127, 128, 129, 130 and upregulate155, 156 pro-inflammatory signaling. PI3K consists of multiple subunits,157 which could contribute to these observed differences,124, 130, 158, 159 as could differential strength of PI3K signaling induced by distinct PRR. Furthermore, the subunits targeted vary between different pharmacological PI3K inhibitors and the inhibitor concentrations used. In human disease, targeting the PI3K substrate mTOR (mammalian target of rapamycin) induces tolerance during transplantation by upregulating regulatory T cells.148 However, rapamycin inhibits PRR-mediated tolerance in human macrophages,126 and mTORC1 deficiency in DCs exacerbates DSS-induced colitis.160 Therefore, although PI3K and mTOR signaling can negatively regulate PRR-initiated inflammatory pathways in macrophages, this signaling in T cells can lead to distinct outcomes.
microRNAs inhibit PRR signaling on a transcriptional level
A rapidly developing field of PRR-signaling regulation is microRNAs (Figure 1). MicroRNAs regulate inflammation through targeting the 3' untranslated region of transcripts leading to either their stabilization or degradation.161 Such regulation is central to fine-tuning inflammatory responses. Interestingly, microRNAs can modify expression levels of both the positive and negative PRR-signaling regulators, including MyD88 signaling intermediates, transcription factors, cytokines, and inhibitors. Multiple microRNAs have been associated with TLR signaling,161 with miR-146 and miR-155 being prominent in regulating inflammation.161 For example, miR-146 inhibits IRAK-1 expression in human macrophages, thereby decreasing NF-κB activation and pro-inflammatory cytokine induction.162 Polymorphisms altering miR-146 expression are associated with SLE and RA.163 Similarly, miR-23b, which suppresses multiple pro-inflammatory molecules, ameliorates mouse lupus, RA, and MS models and is downregulated in human autoimmune diseases, including RA.164 By contrast, microRNAs such as miR-155 that downregulate PRR-signaling inhibitors are increased in macrophages from RA patients. Consistently, miR-155-deficient mice are protected against experimental arthritis.165 Interestingly, microRNAs can act as TLR7 and TLR9 agonists, inducing paracrine inflammatory responses.166 Multiple human inflammatory diseases, including UC167 and RA,168 show dysregulated microRNA profiles. Expression and function of microRNAs can vary in human and mouse immune cells;169 elucidating these differences is crucial to understanding the mechanisms through which distinct microRNAs regulate separate human macrophage functions.
Epigenetic regulation of PRR signaling
Immune cell phenotypes can be broadly modulated by epigenetic modifications (Figure 1). Upon PRR restimulation of mouse and human macrophages following chronic LPS stimulation, some genes are repressed (“tolerant”), whereas others remain transcribed (“non-tolerant”);16, 17 such outcomes are partially regulated through epigenetic modifications. The tolerant genes largely include inflammatory genes, while the non-tolerant genes mediate microbial killing and inhibit inflammation.16 Importantly, in mucosal tissues such as the intestine, which chronically encounter microbial products, the epigenetic regulation mediating this dual transcriptional and functional regulation may ultimately enhance anti-microbial function while minimizing tissue inflammation and injury. In mouse lamina propria myeloid cells, bacterial-induced IL-10 was found to downregulate IL-12p40 through histone deacetylase 3 (HDAC3)-mediated histone deacetylation.170 However, overall epigenetic regulation of intestinal macrophages is poorly defined. In primary human DCs and THP-1 macrophages, acute LPS stimulation results in histone acetylation and H3K4 methylation (both activating transcription) in pro-inflammatory cytokine and activation marker genes.171, 172 However, during endotoxin tolerance, inhibitory histone methylation (e.g., H3K27) is observed on pro-inflammatory genes in these cells.172, 173, 174 Interestingly, a polymorphism in TLE1, a HDAC-interacting transcription factor, is associated with CD.175 On the other hand, HDAC inhibitors improve experimental colitis and kidney disease in mice176, 177 and are undergoing evaluation as therapy for inflammatory diseases, such as systemic onset juvenile idiopathic arthritis178 and IBD.179 The therapeutic properties of HDAC inhibitors are incompletely understood and may involve mechanisms independent of inflammatory gene regulation, including protein acetylation and the induction of apoptosis.180
Inflammasomes regulate PRR-induced inflammation and are associated with human inflammatory diseases
Microbial stimuli can activate inflammasomes, macromolecular complexes containing NLR family members activating caspase-1, which cleaves pro-IL-1β and -IL-18 into their active forms (Figure 1).181 Multiple NLRs contribute to inflammasome diversity,181 although not all NLR proteins form inflammasomes. Tight balance of inflammasome regulation is crucial; inhibition or deletion of inflammasome components can either ameliorate or exacerbate animal inflammatory disease models. For example, ASC-deficient mice are more resistant to endotoxin shock,182 and caspase-1 deficiency attenuates DSS colitis.183 However, NLRC4 and NLRP6 deficiency exacerbates experimental mouse colitis, partially through dysregulated inflammatory cytokines, including IL-18, and altered intestinal microbiota.184, 185 Furthermore, polymorphisms in NLRs and molecules regulating inflammasome activation are associated with the autoinflammatory diseases.186, 187, 188 Inflammasome-associated pathways also mediate mucosal-associated inflammatory diseases as evidenced by the association of NLRP1 to vitiligo and systemic sclerosis,189, 190 NOD2 to CD,191, 192 and CARD9, IL-18RAP/IL12RL2/IL18R1/IL1RL1 and IL1R2 regions to IBD.10, 59 Unlike the activating role of many NLRs, most, but not all,193 reports find that NLRP12 inhibits inflammation.194, 195 Consistently, NLRP12-deficient mice are more susceptible to DSS-induced colitis and colitis-associated tumorigenesis,196 although not to airway hypersensitivity.197 NLRP12 loss-of-function mutations are associated with inflammatory diseases,198, 199 including atopic dermatitis.200 Studies defining diseases that benefit from targeting either the inflammasome directly or the products of inflammasome activation (e.g., IL-1, IL-18) are ongoing.201
Autophagy regulates multiple macrophage functions
PRR stimulation induces autophagy, which facilitates cellular organelle and bacterial clearance.202 In vivo mouse studies and human genetic association studies have demonstrated that autophagy-associated genes and pathways are essential for intestinal homeostasis;203, 204, 205, 206, 207 loss-of-function polymorphisms in the autophagy genes ATG16L1 and IRGM are associated with CD.10, 208 These polymorphisms impair bacterial killing in some,203, 207, 209 but not all, situations.207, 210 ATG16L1 hypomorphic mice have dysregulated Paneth cell morphology and exhibit microbiota-driven intestinal inflammation.205, 211 Moreover, ATG16L1 can contribute to anti-viral activity in macrophages in an autophagy-independent manner.212 Interestingly, besides mediating bacterial killing, autophagy downregulates cytokine production from myeloid cells. Recent mouse studies demonstrate that autophagy promotes absent in melanoma 2 (AIM2) and NLRP3 inflammasome degradation213 and decreases IL-1 secretion.214, 215 Human peripheral blood mononuclear cells studies show that autophagy also downregulates IL-1 through degrading IL-l transcripts.216 Therefore, autophagy regulates at least two distinct and critical PRR-mediated functions: (1) microbial clearance and (2) cytokine downregulation (Figure 1). These dual functions are crucial in the intestine, thereby highlighting fundamental mechanisms through which autophagy can contribute to intestinal immune homeostasis.
PRR Signaling in Intestinal Macrophages is Mediated by Diverse Local Factors
In previous sections, we discussed mechanisms inhibiting PRR-mediated functions and emphasized in select places how some of these mechanisms contribute to intestinal macrophage function and intestinal homeostasis; here we focus specifically on aspects of the unique phenotype observed in intestinal macrophages and on additional factors and mechanisms contributing to this phenotype. Intestinal macrophages constitute one of the largest reservoirs of myeloid cells.25, 217 Macrophages are located throughout the intestinal tract,25, 218 but most prominently in the lamina propria, beneath the protective epithelial layer, making macrophages particularly important in bacterial recognition following bacterial translocation during events such as epithelial injury.25 Relative to peripheral monocyte-derived cells, intestinal macrophages secrete low levels of pro-inflammatory cytokines upon PRR stimulation but upregulate bacterial killing.136, 219, 220 This limits unnecessary inflammation and tissue damage while simultaneously protecting against overgrowth of resident microbiota and pathogenic bacteria. As peripheral monocytes enter the intestinal lamina propria, multiple local mechanisms contribute to their differentiation into intestinal macrophages.25 Contributing factors include microbial components (e.g., PRR ligands, polysaccharide A221), anti-inflammatory mediators (e.g., TGF-β, IL-10), nutrients, and apoptotic cells. Notably, during acute infection or tissue injury, intestinal macrophages can mount inflammatory responses.220 This inflammation can be mediated by peripheral monocyte recruitment to the intestine; the intestinal microenvironment can then influence subsequent differentiation patterns.222, 223, 224, 225, 226, 227 Consistently, altered proportions, functions, and/or differentiation of intestinal myeloid cells can lead to intestinal inflammation in mice,222, 228, 229, 230, 231, 232, 233, 234, 235, 236 and myeloid cell dysregulation in human intestine is observed in IBD.237, 238
Consistent with decreased pro-inflammatory cytokine secretion, intestinal macrophages demonstrate downregulated CD14, MD2, TLR2, TLR4, MyD88, and IRAK-1, and decreased NF-κB activation, although certain PRR (e.g., TLR3, 5–9) are expressed.26, 136 Chronic PRR stimulation could contribute to downregulated inflammation in intestinal macrophages. For example, chronic NOD2 stimulation downregulates cytokines in myeloid-derived cells31, 102, 126 and protects mice from experimental colitis.102 Interestingly, intestinal myeloid cells from germ-free mice show downregulated PRR-mediated cytokine secretion,170, 220 although these mice were exposed to PRR ligands through food and bedding. To clearly dissect the role of chronic PRR stimulation in the downregulated cytokines observed in intestinal macrophages, germ-free mice will need to be examined under conditions of food and bedding devoid of microbial products, combined with recurrent intestinal injury. Stromal and epithelial cell secretions (e.g., TGF-β, IL-10, retinoic acid, and thymic stromal lymphopoietin) can also downregulate intestinal myeloid cell responses.26, 136, 239, 240, 241, 242 For example, TGF-β signaling in DCs regulates intestinal inflammation in mice,243 and intestinal stromal cell-derived TGF-β downregulates CD14 expression, NF-κB activation, and PRR-induced cytokine secretion in peripheral human monocytes.26, 136, 240 Nutrients, including vitamin D and retinoic acid that are abundant in the intestine, also contribute to intestinal macrophage tolerance. Vitamin D downregulates PRR-induced pro-inflammatory cytokines from human monocytes.244 Consistently, vitamin D administration attenuates experimental mouse colitis.245, 246 Furthermore, higher vitamin D plasma levels correlate to decreased CD risk.247 Conversely, compared with healthy controls, CD patients exhibit vitamin D deficiency,248 which is multi-factorial in etiology, and correlates to disease severity.249 Similarly, in addition to its immunoregulatory roles in T cells,250 retinoic acid downregulates pro-inflammatory cytokines in PRR-stimulated human DC.251 Finally, intestinal macrophages ingest apoptotic cells, which leads to TGF-β and prostaglandin production.252, 253, 254 Importantly, mice lacking intermediates in apoptosis-inducing pathways, such as TAM receptors, C1q, MFG-E8, and TIM-4, develop autoimmunity.86, 255, 256, 257 Moreover, autophagy clears apoptotic debris,258 highlighting one mechanism through which autophagy dysfunction may contribute to IBD susceptibility.203, 204, 205, 206, 207
Despite downregulated cytokines, human intestinal macrophages upregulate bactericidal activity. Intestinal factors can mediate both processes. PRR16, 17 and Vitamin D259, 260 stimulation of human and mouse macrophages upregulates multiple anti-microbial pathways. Interestingly, some studies show bactericidal defects in CD patient macrophages.261 As heterogeneous mechanisms lead to CD, bactericidal defects likely exist in a subset of CD patients, such as those carrying polymorphisms in bactericidal pathways (e.g., NOD2, ATG16L1, IRGM, and NCF-2).10, 262 Targeting mechanisms mediating the dichotomy of downregulated inflammatory and upregulated bactericidal pathways in human intestinal macrophages might be particularly beneficial in IBD therapy.
Human and Mouse Macrophages Demonstrate Distinct Regulation in Various Pathways
Human and murine-based studies are essential and complementary to defining mechanisms of disease pathogenesis. There are multiple, fundamental differences between human and mouse PRR signaling outcomes,11, 12 including distinct PRR and cytokine stimulation responsivity,31, 102, 263, 264, 265, 266, 267 and differential PRR utilization.268 These differences are critical when extending mouse findings to human physiology and disease. Reasons for inter-species differences include distinct function and/or expression of relevant genes, dissimilar life spans, distinct microbial colonization, and altered environmental exposures. Furthermore, human genetic diversity is greater than that of experimental inbred mice, adding significant complexity, but also unique opportunities, in pursuing human immunology.
Different factors regulate mouse and human macrophage polarization
As macrophages enter or are activated in various microenvironments, they differentiate into distinct subtypes, characterized by differential surface marker, cytokine, and protein expression. Two broad categories of polarized mouse macrophages include classically activated macrophages (M1), associated with pro-inflammatory cytokine secretion, and alternatively activated macrophages (M2), associated with an anti-inflammatory phenotype.6 Distinct factors mediate mouse and human macrophage polarization and macrophage phenotypes. M1- and M2-like human macrophages exist269, 270 but are less well defined than their mouse counterparts. IFN-γ stimulation of mouse, but not human M1 macrophages, dramatically induces NOS2, and IL-4-stimulated human macrophages produce significantly less arginase than mouse M2 macrophages.270 Human macrophage polarization likely involves specific transcription factors. Although in one study, IRF5 mediated M1, and IRF4 mediated M2 polarization of human macrophages,269 another study found contrasting results.271 Notably, IRF5 is central to macrophage function as evidenced by IRF5 polymorphism associations with numerous autoimmune/inflammatory diseases exhibiting dysregulated cytokine expression272 and the dramatic contribution of IRF5 polymorphisms to human variance in PRR-induced cytokine secretion.273 Another IRF family member, IRF8, has been recently shown to promote mouse M1 polarization;274 IRF8 effects on human macrophage polarization are still unclear. Further studies are needed to better define the regulation of human macrophage polarization and how this polarization contributes to immune homeostasis.
Mouse and human macrophages demonstrate differences in inflammasome activation
PRR stimulation induces pro-IL-1β, but a second signal, such as adenosine triphosphate (ATP), is required to activate P2 × 7 receptors. This results in potassium efflux and calcium influx, which activates the inflammasome and induces caspase-1-mediated processing of pro-IL-1β and -IL-18 to their active forms.275, 276, 277 Tissue damage releases ATP,278 thereby providing a second signal to mouse macrophages.279 However, PRR-ligand stimulated human monocytes secrete autocrine ATP;280 therefore, PRR-stimulation alone induces IL-1β in these cells. Some studies demonstrate that PRR-stimulated human macrophages do not secrete IL-1β,281, 282 with a second stimulus necessary for IL-1β secretion.283 However, others detect active IL-1β in PRR-stimulated human macrophages due to the sufficiency of PRR stimulation for ATP production.140, 153, 284, 285, 286 These differences might partially reflect different culture conditions used to generate macrophages; growth factor and cytokine differences can profoundly influence human macrophage responses.287 Improved insight into differential regulation of IL-1 in human and mouse macrophages will be important, as IL-1 contributes to multiple human diseases,201 and autocrine/paracrine IL-1β dramatically amplifies PRR-induced cytokine secretion in human macrophages.140, 153
Human variance adds complexity to examination of PRR-induced macrophage functions
Mouse studies minimize variance in PRR-induced inflammatory outcomes through inbreeding, housing in conditions that reduce environmental differences, and utilizing age-, gender-, and littermate-matched mice. However, humans show significant variance in their genetic background,288 environmental exposures,289, 290 and intestinal microbiota,291, 292, 293, 294 which translates into broad inter-individual immunological differences. For example, there is dramatic inter-individual variation in cytokine and inhibitor molecule induction upon PRR stimulation in human macrophages.31, 61, 273, 295, 296, 297, 298 Such variance likely affects the balance between susceptibility to infections versus autoimmune/inflammatory diseases. Genes identified to regulate variance in human cytokine secretion include IRF5, IL-1Ra, and TLR1. IRF5 polymorphisms account for up to 53% of variance in PRR-induced TNF-α secretion from human monocyte-derived cells;273 this dramatic contribution likely results from the distinct genotypes being commonly distributed across the population and from the dramatic gene-dose-dependent regulation mediated by IRF5 polymorphisms. IL-1Ra polymorphisms mediate 5% of variance in constitutive IL-1β plasma levels,299 and TLR1 polymorphisms contribute to variance in IL-6 secretion during sepsis.297 Perhaps not coincidentally, these polymorphisms contribute to susceptibility and/or outcomes in multiple inflammatory/autoimmune diseases associated with dysregulated cytokine production.272, 297, 299 Importantly, inhibitory mechanisms regulating PRR-initiated pathways demonstrate varying contributions in myeloid cells from different individuals (e.g., IRAK-M and SHIP-1).31, 61 Of note is that mutations modulating inflammatory pathway intermediates, such as MyD88300 and TRIF,301 may dramatically affect cytokine induction, but if relatively rare, will not significantly influence overall inter-individual heterogeneity. Another consideration is that some PRR-pathway polymorphisms (e.g., NOD2,302, 303 TLR4304), have different frequencies across distinct ancestries, which will therefore influence the inter-individual differences upon PRR stimulation between population groups. Host–microbe interactions are central in natural selection and functional variation; the inter-individual variability inherent in human immunological studies can ultimately be leveraged to define underlying mechanisms of autoimmune-mediated diseases.305
Another contribution to variance in human immune responses is inter-individual differences in microbial composition. Multiple mouse studies have implicated intestinal microbiota in regulating immunity and disease development, including mucosal diseases.185, 221, 306, 307, 308 Altered human intestinal microbial composition is also associated with dysregulated immunity and disease;291, 292, 293, 294 it is unclear to what degree these microbial changes are a consequence or a cause of the inter-individual immunological differences.
Taken together, mouse models have provided tremendous insight into defining the importance of various pathways and functions in myeloid-derived cells in health and in disease development. Furthermore, humanized mice can be used to address select in vivo myeloid cell functions given successful reconstitution of human myeloid cells into mice.309 However, in applying the information from mouse studies to human immune function, it is critical to understand the similarities and differences between mouse and human immune pathways.
Future Perspectives
Despite significant advances in understanding human macrophage regulation and functions, multiple questions remain. What are the functional outcomes of the many disease-associated polymorphisms in macrophages? Which factors influence human macrophage polarization? How do monocytes acquire distinct resident phenotypes as they enter tissues? How do intestinal macrophages determine when to maintain tolerance and when to mount inflammatory responses to resident or pathogenic bacteria?
Human heterogeneity poses a specific challenge when conducting human macrophage studies; sampling from well-powered cohorts is essential. Moreover, limited tissue access restricts the number of functional immunological readouts; high throughput approaches minimizing sample sizes are continuously being developed and improved. Uniform sample processing, standard operating techniques and normalization of immune readouts based on criteria, including age, gender, ancestry, or specific genotypes, will be essential for future studies. Despite these challenges, elucidating the inhibitory mechanisms in primary human macrophages is essential to fully understand mechanisms mediating both health and disease.
References
Abraham, C. & Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 140, 1729–1737 (2011).
Chen, Y., Lui, V.C., Rooijen, N.V. & Tam, P.K. Depletion of intestinal resident macrophages prevents ischaemia reperfusion injury in gut. Gut 53, 1772–1780 (2004).
Hunter, M.M. et al. In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 138, 1395–1405 (2010).
McGaha, T.L., Chen, Y., Ravishankar, B., van Rooijen, N. & Karlsson, M.C. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 117, 5403–5412 (2011).
Rugtveit, J. et al. Respiratory burst of intestinal macrophages in inflammatory bowel disease is mainly caused by CD14+L1+ monocyte derived cells. Gut 37, 367–373 (1995).
Lawrence, T. & Natoli, G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 (2011).
Hume, D.A. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J. Leukoc. Biol. 89, 525–538 (2011).
Rakoff-Nahoum, S. & Medzhitov, R. Innate immune recognition of the indigenous microbial flora. Mucosal Immunol. 1 (Suppl 1), S10–S14 (2008).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 (2004).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Mestas, J. & Hughes, C.C. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004).
Davis, M.M. A prescription for human immunology. Immunity 29, 835–838 (2008).
Murray, P.J. & Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011).
Fahy, R.J., Doseff, A.I. & Wewers, M.D. Spontaneous human monocyte apoptosis utilizes a caspase-3-dependent pathway that is blocked by endotoxin and is independent of caspase-1. J. Immunol. 163, 1755–1762 (1999).
Becker, S., Warren, M.K. & Haskill, S. Colony-stimulating factor-induced monocyte survival and differentiation into macrophages in serum-free cultures. J. Immunol. 139, 3703–3709 (1987).
Foster, S.L., Hargreaves, D.C. & Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978 (2007).
Pena, O.M., Pistolic, J., Raj, D., Fjell, C.D. & Hancock, R.E. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J. Immunol. 186, 7243–7254 (2011).
Randow, F. et al. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J. Exp. Med. 181, 1887–1892 (1995).
Fan, H. & Cook, J.A. Molecular mechanisms of endotoxin tolerance. J. Endotoxin. Res. 10, 71–84 (2004).
Medvedev, A.E., Lentschat, A., Wahl, L.M., Golenbock, D.T. & Vogel, S.N. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J. Immunol. 169, 5209–5216 (2002).
Medvedev, A.E., Kopydlowski, K.M. & Vogel, S.N. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J. Immunol. 164, 5564–5574 (2000).
Nomura, F. et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 164, 3476–3479 (2000).
Adib-Conquy, M. & Cavaillon, J.M. Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to MyD88 and not by modulating TLR4 expression. J. Biol. Chem. 277, 27927–27934 (2002).
Medvedev, A.E. et al. Induction of tolerance to lipopolysaccharide and mycobacterial components in Chinese hamster ovary/CD14 cells is not affected by overexpression of Toll-like receptors 2 or 4. J. Immunol. 167, 2257–2267 (2001).
Smith, P.D. et al. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. 4, 31–42 (2011).
Smythies, L.E. et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J. Biol. Chem. 285, 19593–19604 (2010).
Cario, E. & Podolsky, D.K. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 68, 7010–7017 (2000).
Fukata, M. et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133, 1869–1881 (2007).
Fukata, M. et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm. Bowel Dis. 17, 1464–1473 (2010).
Vig, E. et al. Modulation of tumor necrosis factor and interleukin-1-dependent NF-kappaB activity by mPLK/IRAK. J. Biol. Chem. 274, 13077–13084 (1999).
Hedl, M., Li, J., Cho, J.H. & Abraham, C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc. Natl. Acad. Sci. USA 104, 19440–19445 (2007).
Lotz, M. et al. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 203, 973–984 (2006).
Suzuki, N. et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416, 750–756 (2002).
Toubiana, J. et al. IRAK1 functional genetic variant affects severity of septic shock. Crit. Care Med. 38, 2287–2294 (2010).
Arcaroli, J. et al. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am. J. Respir. Crit. Care Med. 173, 1335–1341 (2006).
Picard, C. et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89, 403–425 (2010).
Sha, W.C., Liou, H.C., Tuomanen, E.I. & Baltimore, D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 80, 321–330 (1995).
Kang, S.M., Tran, A.C., Grilli, M. & Lenardo, M.J. NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science 256, 1452–1456 (1992).
Oeckinghaus, A., Hayden, M.S. & Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 12, 695–708 (2011).
Tomczak, M.F. et al. NF-kappa B is required within the innate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J. Immunol. 171, 1484–1492 (2003).
Bohuslav, J. et al. Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J. Clin. Invest. 102, 1645–1652 (1998).
Porta, C. et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 106, 14978–14983 (2009).
Greten, F.R. et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130, 918–931 (2007).
Erdman, S., Fox, J.G., Dangler, C.A., Feldman, D. & Horwitz, B.H. Typhlocolitis in NF-kappa B-deficient mice. J. Immunol. 166, 1443–1447 (2001).
Neurath, M.F., Pettersson, S., Meyer zum Buschenfelde, K.H. & Strober, W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat. Med. 2, 998–1004 (1996).
Guma, M. et al. Constitutive intestinal NF-kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. J. Exp. Med. 208, 1889–1900 (2011).
Ishikawa, H. et al. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-kappaB1) but expressing p50. J. Exp. Med. 187, 985–996 (1998).
Sampath, V. et al. The NFKB1 (g.-24519delATTG) variant is associated with necrotizing enterocolitis (NEC) in premature infants. J. Surg. Res. 169, e51–e57 (2011).
Neurath, M.F. et al. Cytokine gene transcription by NF-kappa B family members in patients with inflammatory bowel disease. Ann. NY Acad. Sci. 859, 149–159 (1998).
Rogler, G. et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 115, 357–369 (1998).
Litvak, V. et al. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 10, 437–443 (2009).
Brint, E.K. et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat. Immunol. 5, 373–379 (2004).
Sweet, M.J. et al. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J. Immunol. 166, 6633–6639 (2001).
Takezako, N. et al. ST2 suppresses IL-6 production via the inhibition of IkappaB degradation induced by the LPS signal in THP-1 cells. Biochem. Biophys. Res. Commun. 341, 425–432 (2006).
Dinarello, C.A. An IL-1 family member requires caspase-1 processing and signals through the ST2 receptor. Immunity 23, 461–462 (2005).
Pastorelli, L. et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl. Acad. Sci. USA 107, 8017–8022 (2010).
Coyle, A.J. et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J. Exp. Med. 190, 895–902 (1999).
Seidelin, J.B., Rogler, G. & Nielsen, O.H. A role for interleukin-33 in T(H)2-polarized intestinal inflammation? Mucosal Immunol. 4, 496–502 (2011).
Franke, A. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 42, 1118–1125 (2010).
Kobayashi, K. et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110, 191–202 (2002).
Park, S.H., Park-Min, K.H., Chen, J., Hu, X. & Ivashkiv, L.B. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat. Immunol. 12, 607–615 (2011).
del Fresno, C. et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J. Immunol. 182, 6494–6507 (2009).
van’t Veer, C. et al. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J. Immunol. 179, 7110–7120 (2007).
Seki, M. et al. Critical role of IL-1 receptor-associated kinase-M in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J. Immunol. 184, 1410–1418 (2010).
Biswas, A. et al. Negative regulation of Toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur. J. Immunol. 41, 182–194 (2011).
Balaci, L. et al. IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am. J. Hum. Genet. 80, 1103–1114 (2007).
Nakashima, K. et al. An association study of asthma and related phenotypes with polymorphisms in negative regulator molecules of the TLR signaling pathway. J. Hum. Genet. 51, 284–291 (2006).
Nanthakumar, N. et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS One 6, e17776 (2011).
Almeida, A.S. et al. Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J. Immunol. 183, 718–731 (2009).
Endo, T.A. et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387, 921–924 (1997).
Starr, R. et al. A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921 (1997).
Nakagawa, R. et al. SOCS-1 participates in negative regulation of LPS responses. Immunity 17, 677–687 (2002).
Alexander, W.S. et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98, 597–608 (1999).
Prele, C.M. et al. SOCS1 regulates the IFN but not NFkappaB pathway in TLR-stimulated human monocytes and macrophages. J. Immunol. 181, 8018–8026 (2008).
Kinjyo, I. et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 17, 583–591 (2002).
Baetz, A., Frey, M., Heeg, K. & Dalpke, A.H. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J. Biol. Chem. 279, 54708–54715 (2004).
Gingras, S., Parganas, E., de Pauw, A., Ihle, J.N. & Murray, P.J. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J. Biol. Chem. 279, 54702–54707 (2004).
Mostecki, J. et al. A SOCS-1 promoter variant is associated with total serum IgE levels. J. Immunol. 187, 2794–2802 (2011).
Harada, M. et al. Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am. J. Respir. Cell Mol. Biol. 36, 491–496 (2007).
Rothlin, C.V., Ghosh, S., Zuniga, E.I., Oldstone, M.B. & Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136 (2007).
Lu, Q. & Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293, 306–311 (2001).
Sharif, M.N. et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med. 203, 1891–1901 (2006).
Camenisch, T.D., Koller, B.H., Earp, H.S. & Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 162, 3498–3503 (1999).
Scutera, S. et al. Survival and migration of human dendritic cells are regulated by an IFN-alpha-inducible Axl/Gas6 pathway. J. Immunol. 183, 3004–3013 (2009).
Cohen, P.L. et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 196, 135–140 (2002).
Scott, R.S. et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 (2001).
Seitz, H.M., Camenisch, T.D., Lemke, G., Earp, H.S. & Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 178, 5635–5642 (2007).
Ravichandran, K.S. & Lorenz, U. Engulfment of apoptotic cells: signals for a good meal. Nat. Rev. Immunol. 7, 964–974 (2007).
Aadland, E., Odegaard, O.R., Roseth, A. & Try, K. Free protein S deficiency in patients with chronic inflammatory bowel disease. Scand. J. Gastroenterol. 27, 957–960 (1992).
Brouwer, J.L., Bijl, M., Veeger, N.J., Kluin-Nelemans, H.C. & van der Meer, J. The contribution of inherited and acquired thrombophilic defects, alone or combined with antiphospholipid antibodies, to venous and arterial thromboembolism in patients with systemic lupus erythematosus. Blood 104, 143–148 (2004).
Lee, E.G. et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289, 2350–2354 (2000).
Wertz, I.E. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699 (2004).
Boone, D.L. et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5, 1052–1060 (2004).
Saitoh, T. et al. A20 is a negative regulator of IFN regulatory factor 3 signaling. J. Immunol. 174, 1507–1512 (2005).
Hitotsumatsu, O. et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 28, 381–390 (2008).
Hammer, G.E. et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat. Immunol. 12, 1184–1193 (2011).
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Musone, S.L. et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat. Genet. 40, 1062–1064 (2008).
Graham, R.R. et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 40, 1059–1061 (2008).
Thomson, W. et al. Rheumatoid arthritis association at 6q23. Nat. Genet. 39, 1431–1433 (2007).
Plenge, R.M. et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat. Genet. 39, 1477–1482 (2007).
Watanabe, T. et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J. Clin. Invest. 118, 545–559 (2008).
Negishi, H. et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. USA 102, 15989–15994 (2005).
Honma, K. et al. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc. Natl. Acad. Sci. USA 102, 16001–16006 (2005).
Mudter, J. et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J. Clin. Invest. 118, 2415–2426 (2008).
Gabhann, J.N. et al. Absence of SHIP-1 results in constitutive phosphorylation of tank-binding kinase 1 and enhanced TLR3-dependent IFN-beta production. J. Immunol. 184, 2314–2320 (2010).
Sly, L.M., Rauh, M.J., Kalesnikoff, J., Song, C.H. & Krystal, G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity 21, 227–239 (2004).
Rauh, M.J. et al. SHIP represses the generation of alternatively activated macrophages. Immunity 23, 361–374 (2005).
Kerr, W.G., Park, M.Y., Maubert, M. & Engelman, R.W. SHIP deficiency causes Crohn’s disease-like ileitis. Gut 60, 177–188 (2011).
Helgason, C.D. et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 12, 1610–1620 (1998).
Arijs, I. et al. Intestinal expression of SHIP in inflammatory bowel diseases. Gut 61, 956–957 (2011).
Wang, L. et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity 32, 518–530 (2010).
Kanamaru, Y. et al. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J. Immunol. 180, 2669–2678 (2008).
Leon, F. et al. Antibodies to complement receptor 3 treat established inflammation in murine models of colitis and a novel model of psoriasiform dermatitis. J. Immunol. 177, 6974–6982 (2006).
Bogdan, C., Paik, J., Vodovotz, Y. & Nathan, C. Contrasting mechanisms for suppression of macrophage cytokine release by transforming growth factor-beta and interleukin-10. J. Biol. Chem. 267, 23301–23308 (1992).
Takeuchi, M., Alard, P. & Streilein, J.W. TGF-beta promotes immune deviation by altering accessory signals of antigen-presenting cells. J. Immunol. 160, 1589–1597 (1998).
Murray, P.J. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc. Natl. Acad. Sci. USA 102, 8686–8691 (2005).
Kontoyiannis, D. et al. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. Embo J. 20, 3760–3770 (2001).
Chan, C.S. et al. Interleukin-10 inhibits LPS induced TNFalpha translation through a SHIP1-dependent pathway. J. Biol. Chem. 287, 38020–38027 (2012).
Chang, J., Kunkel, S.L. & Chang, C.H. Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc. Natl. Acad. Sci. USA 106, 18327–18332 (2009).
Lang, R., Patel, D., Morris, J.J., Rutschman, R.L. & Murray, P.J. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 169, 2253–2263 (2002).
Williams, L.M., Ricchetti, G., Sarma, U., Smallie, T. & Foxwell, B.M. Interleukin-10 suppression of myeloid cell activation--a continuing puzzle. Immunology 113, 281–292 (2004).
Sabat, R. et al. Biology of interleukin-10. Cytokine Growth Factor Rev. 21, 331–344 (2010).
Uno, J.K. et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110delta. Gastroenterology 139, 1642–1653 1653 e1-6 (2010).
Chaurasia, B. et al. Phosphoinositide-dependent kinase 1 provides negative feedback inhibition to Toll-like receptor-mediated NF-kappaB activation in macrophages. Mol. Cell Biol. 30, 4354–4366 (2010).
Hedl, M. & Abraham, C. Secretory mediators regulate Nod2-induced tolerance in human macrophages. Gastroenterology 140, 231–241 (2011).
Guha, M. & Mackman, N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J. Biol. Chem. 277, 32124–32132 (2002).
Fukao, T. et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3, 875–881 (2002).
Martin, M. et al. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J. Immunol. 171, 717–725 (2003).
Gonzalez-Garcia, A., Sanchez-Ruiz, J., Flores, J.M. & Carrera, A.C. Phosphatidylinositol 3-kinase gamma inhibition ameliorates inflammation and tumor growth in a model of colitis-associated cancer. Gastroenterology 138, 1374–1383 (2010).
Jarry, A. et al. Mucosal IL-10 and TGF-beta play crucial roles in preventing LPS-driven, IFN-gamma-mediated epithelial damage in human colon explants. J. Clin. Invest. 118, 1132–1142 (2008).
Elson, C.O. et al. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol. Rev. 206, 260–276 (2005).
Watanabe, N. et al. Elimination of local macrophages in intestine prevents chronic colitis in interleukin-10-deficient mice. Dig. Dis. Sci. 48, 408–414 (2003).
Murai, M. et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178–1184 (2009).
Rani, R., Smulian, A.G., Greaves, D.R., Hogan, S.P. & Herbert, D.R. TGF-beta limits IL-33 production and promotes the resolution of colitis through regulation of macrophage function. Eur. J. Immunol. 41, 2000–2009 (2011).
Smythies, L.E. et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Invest. 115, 66–75 (2005).
Glocker, E.O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009).
Glocker, E.O. et al. Infant colitis-it’s in the genes. Lancet 376, 1272 (2010).
Gabay, C., Lamacchia, C. & Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 6, 232–241 (2010).
Hedl, M. & Abraham, C. Distinct roles for Nod2 protein and autocrine interleukin-1{beta} in muramyl dipeptide-induced mitogen-activated protein kinase activation and cytokine secretion in human macrophages. J. Biol. Chem. 286, 26440–26449 (2011).
Ferretti, M. et al. Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J. Clin. Invest. 94, 449–453 (1994).
Hirsch, E., Irikura, V.M., Paul, S.M. & Hirsh, D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. USA 93, 11008–11013 (1996).
Kastner, D.L., Aksentijevich, I. & Goldbach-Mansky, R. Autoinflammatory disease reloaded: a clinical perspective. Cell 140, 784–790 (2011).
Katakura, K. et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Invest. 115, 695–702 (2005).
Kollias, G., Douni, E., Kassiotis, G. & Kontoyiannis, D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol. Rev. 169, 175–194 (1999).
Jacob, C.O. & McDevitt, H.O. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature 331, 356–358 (1988).
Hommes, D. et al. Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease. Gastroenterology 122, 7–14 (2002).
Roncarolo, M.G. & Battaglia, M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat. Rev. Immunol. 7, 585–598 (2007).
Yi, A.K. et al. Role of mitogen-activated protein kinases in CpG DNA-mediated IL-10 and IL-12 production: central role of extracellular signal-regulated kinase in the negative feedback loop of the CpG DNA-mediated Th1 response. J. Immunol. 168, 4711–4720 (2002).
Lucas, M., Zhang, X., Prasanna, V. & Mosser, D.M. ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL-10 locus. J. Immunol. 175, 469–477 (2005).
Foey, A.D. et al. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-alpha: role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 160, 920–928 (1998).
Ma, W. et al. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J. Biol. Chem. 276, 13664–13674 (2001).
Hedl, M. & Abraham, C. Nod2-induced autocrine interleukin-1 alters signaling by ERK and p38 to differentially regulate secretion of inflammatory cytokines. Gastroenterology 143, 1530–1543 (2012).
Papoutsopoulou, S. et al. ABIN-2 is required for optimal activation of Erk MAP kinase in innate immune responses. Nat. Immunol. 7, 606–615 (2006).
Strassheim, D. et al. Phosphoinositide 3-kinase and Akt occupy central roles in inflammatory responses of Toll-like receptor 2-stimulated neutrophils. J. Immunol. 172, 5727–5733 (2004).
Ojaniemi, M. et al. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur. J. Immunol. 33, 597–605 (2003).
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341 (2010).
Barber, D.F. et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 11, 933–935 (2005).
Camps, M. et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 11, 936–943 (2005).
Ohtani, M. et al. Cutting edge: mTORC1 in intestinal CD11c+CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. 188, 4736–4740 (2012).
O’Neill, L.A., Sheedy, F.J. & McCoy, C.E. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 11, 163–175 (2011).
Taganov, K.D., Boldin, M.P., Chang, K.J. & Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 103, 12481–12486 (2006).
Lofgren, S.E. et al. Genetic association of miRNA-146a with systemic lupus erythematosus in Europeans through decreased expression of the gene. Genes Immun. 13, 268–274 (2012).
Zhu, S. et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-alpha. Nat. Med. 18, 1077–1086 (2011).
Kurowska-Stolarska, M. et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc. Natl. Acad. Sci. USA 108, 11193–11198 (2010).
Fabbri, M. et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 109, E2110–E2116 (2012).
Wu, F. et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology 135, 1624–1635 e24 (2008).
Chatzikyriakidou, A., Voulgari, P.V., Georgiou, I. & Drosos, A.A. miRNAs and related polymorphisms in rheumatoid arthritis susceptibility. Autoimmun. Rev. 11, 636–641 (2011).
Rossi, R.L. et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat. Immunol. 12, 796–803 (2011).
Kobayashi, T. et al. IL-10 Regulates Il12b Expression via Histone Deacetylation: Implications for Intestinal Macrophage Homeostasis. J. Immunol. 189, 1792–1799 (2012).
Yoza, B.K., Hu, J.Y. & McCall, C.E. Inhibition of histone deacetylation enhances endotoxin-stimulated transcription but does not reverse endotoxin tolerance. J. Endotoxin Res. 8, 109–114 (2002).
Huang, Y. et al. Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes Immun. 13, 311–320 (2012).
El Gazzar, M. et al. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol. Cell Biol. 29, 1959–1971 (2009).
Chan, C., Li, L., McCall, C.E. & Yoza, B.K. Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J. Immunol. 175, 461–468 (2005).
Nimmo, E.R. et al. TLE1 modifies the effects of NOD2 in the pathogenesis of Crohn’s disease. Gastroenterology 141, 972–981 e1-2 (2011).
Glauben, R. et al. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J. Immunol. 176, 5015–5022 (2006).
Mishra, N., Reilly, C.M., Brown, D.R., Ruiz, P. & Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 111, 539–552 (2003).
Vojinovic, J. & Damjanov, N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol. Med. 17, 397–403 (2011).
Edwards, A.J. & Pender, S.L. Histone deacetylase inhibitors and their potential role in inflammatory bowel diseases. Biochem. Soc. Trans. 39, 1092–1095 (2011).
Dinarello, C.A., Fossati, G. & Mascagni, P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol. Med. 17, 333–352 (2011).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 (2009).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Siegmund, B., Lehr, H.A., Fantuzzi, G. & Dinarello, C.A. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA 98, 13249–13254 (2001).
Carvalho, F.A. et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 5, 288–298 (2012).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Hoffman, H.M., Mueller, J.L., Broide, D.H., Wanderer, A.A. & Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305 (2001).
Feldmann, J. et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 71, 198–203 (2002).
Consortium, T.I.F. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 90, 797–807 (1997).
Dieude, P. et al. NLRP1 influences the systemic sclerosis phenotype: a new clue for the contribution of innate immunity in systemic sclerosis-related fibrosing alveolitis pathogenesis. Ann. Rheum. Dis. 70, 668–674 (2011).
Jin, Y. et al. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 356, 1216–1225 (2007).
Hugot, J.P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603 (2001).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606 (2001).
Vladimer, G.I. et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107 (2012).
Lich, J.D. et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 178, 1256–1260 (2007).
Williams, K.L. et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J. Biol. Chem. 280, 39914–39924 (2005).
Allen, I.C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 36, 742–754 (2012).
Allen, I.C. et al. Characterization of NLRP12 during the Development of Allergic Airway Disease in Mice. PLoS One 7, e30612 (2012).
Borghini, S. et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 63, 830–839 (2011).
Jeru, I. et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 105, 1614–1619 (2008).
Macaluso, F. et al. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp. Dermatol. 16, 692–698 (2007).
Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 (2011).
Delgado, M. et al. Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 227, 189–202 (2009).
Travassos, L.H. et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62 (2010).
McCarroll, S.A. et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 40, 1107–1112 (2008).
Cadwell, K. et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 (2008).
Cooney, R. et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97 (2010).
Homer, C.R., Richmond, A.L., Rebert, N.A., Achkar, J.P. & McDonald, C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139, 1630–1641 1641 e1-2 (2010).
Parkes, M. et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 39, 830–832 (2007).
Brest, P. et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 43, 242–245 (2011).
Glubb, D.M. et al. NOD2 and ATG16L1 polymorphisms affect monocyte responses in Crohn’s disease. World J. Gastroenterol. 17, 2829–2837 (2011).
Cadwell, K. et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145 (2010).
Hwang, S. et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 11, 397–409 (2012).
Shi, C.S. et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 (2011).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008).
Plantinga, T.S. et al. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 60, 1229–1235 (2011).
Lee, S.H., Starkey, P.M. & Gordon, S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J. Exp. Med. 161, 475–489 (1985).
Mowat, A.M. & Bain, C.C. Mucosal macrophages in intestinal homeostasis and inflammation. J. Innate Immun. 3, 550–564 (2011).
Denning, T.L., Wang, Y.C., Patel, S.R., Williams, I.R. & Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086–1094 (2007).
Franchi, L. et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 13, 449–456 (2012).
Mazmanian, S.K., Round, J.L. & Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 (2008).
Platt, A.M., Bain, C.C., Bordon, Y., Sester, D.P. & Mowat, A.M. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 184, 6843–6854 (2010).
Kim, Y.G. et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34, 769–780 (2011).
Rivollier, A., He, J., Kole, A., Valatas, V. & Kelsall, B.L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 209, 139–155 (2012).
Waddell, A. et al. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J. Immunol. 186, 5993–6003 (2011).
Shi, Y., Evans, J.E. & Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521 (2003).
Bogunovic, M. et al. Origin of the lamina propria dendritic cell network. Immunity 31, 513–525 (2009).
Kamada, N. et al. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J. Immunol. 175, 6900–6908 (2005).
Varol, C. et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 31, 502–512 (2009).
Dunay, I.R. et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29, 306–317 (2008).
Shiloh, M.U. et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10, 29–38 (1999).
Annacker, O. et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061 (2005).
Siddiqui, K.R., Laffont, S. & Powrie, F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity 32, 557–567 (2011).
Manicassamy, S. et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329, 849–853 (2010).
Medina-Contreras, O. et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J. Clin. Invest. 121, 4787–4795 (2011).
Bain, C.C. et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6C(hi) monocyte precursors. Mucosal Immunol. advance online publication, 19 September 2012; doi:10.1038/mi.2012.89 (2012).
Kamada, N. et al. Human CD14+ macrophages in intestinal lamina propria exhibit potent antigen-presenting ability. J. Immunol. 183, 1724–1731 (2009).
Kamada, N. et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J. Clin. Invest. 118, 2269–2280 (2008).
Bimczok, D. et al. Stromal regulation of human gastric dendritic cells restricts the Th1 response to Helicobacter pylori. Gastroenterology 141, 929–938 (2011).
Maheshwari, A. et al. TGF-beta2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 140, 242–253 (2011).
Iliev, I.D., Mileti, E., Matteoli, G., Chieppa, M. & Rescigno, M. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2, 340–350 (2009).
Taylor, B.C. et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J. Exp. Med. 206, 655–667 (2009).
Ramalingam, R. et al. Dendritic cell-specific disruption of TGF-beta receptor II eads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J. Immunol. 189, 3878–3893 (2012).
Sadeghi, K. et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur. J. Immunol. 36, 361–370 (2006).
Alex, P. et al. Clcn5 knockout mice exhibit novel immunomodulatory effects and are more susceptible to dextran sulfate sodium-induced colitis. J. Immunol. 184, 3988–3996 (2010).
Cantorna, M.T., Munsick, C., Bemiss, C. & Mahon, B.D. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J. Nutr. 130, 2648–2652 (2000).
Ananthakrishnan, A.N. et al. Higher Predicted Vitamin D Status Is Associated With Reduced Risk of Crohn’s Disease. Gastroenterology 142, 482–489 (2011).
Raftery, T., O'Morain, C. & O'Sullivan, M. Vitamin D: new Roles and Therapeutic Potential in Inflammatory Bowel Disease. Curr. Drug Metab. 13, 1294–1302 (2012).
Ulitsky, A. et al. Vitamin D deficiency in patients with inflammatory bowel disease: association with disease activity and quality of life. J. Parenter. Enteral. Nutr. 35, 308–316 (2011).
Hall, J.A., Grainger, J.R., Spencer, S.P. & Belkaid, Y. The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22 (2011).
Zapata-Gonzalez, F. et al. 9-cis-Retinoic acid (9cRA), a retinoid X receptor (RXR) ligand, exerts immunosuppressive effects on dendritic cells by RXR-dependent activation: inhibition of peroxisome proliferator-activated receptor gamma blocks some of the 9cRA activities, and precludes them to mature phenotype development. J. Immunol. 178, 6130–6139 (2007).
Voll, R.E. et al. Immunosuppressive effects of apoptotic cells. Nature 390, 350–351 (1997).
Fadok, V.A. et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898 (1998).
Chen, W., Frank, M.E., Jin, W. & Wahl, S.M. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity 14, 715–725 (2001).
Hanayama, R. et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304, 1147–1150 (2004).
Botto, M. et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19, 56–59 (1998).
Rodriguez-Manzanet, R. et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc. Natl. Acad. Sci. USA 107, 8706–8711 (2010).
Kuballa, P., Nolte, W.M., Castoreno, A.B. & Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 30, 611–646 (2012).
Liu, P.T. et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311, 1770–1773 (2006).
Adams, J.S. et al. Vitamin D-directed rheostatic regulation of monocyte antibacterial responses. J. Immunol. 182, 4289–4295 (2009).
Smith, A.M. et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J. Exp. Med. 206, 1883–1897 (2009).
Rioux, J.D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 (2007).
Kim, Y.G. et al. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity 28, 246–257 (2008).
Kapetanovic, R. et al. Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages. Infect. Immun. 75, 830–837 (2007).
Magalhaes, J.G. et al. Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep. 6, 1201–1207 (2005).
Li, J. et al. Regulation of IL-8 and IL-1beta expression in Crohn’s disease associated NOD2/CARD15 mutations. Hum. Mol. Genet. 13, 1715–1725 (2004).
Netea, M.G. et al. The frameshift mutation in Nod2 results in unresponsiveness not only to Nod2- but also Nod1-activating peptidoglycan agonists. J. Biol. Chem. 280, 35859–35867 (2005).
Nahori, M.A. et al. Differential TLR recognition of leptospiral lipid A and lipopolysaccharide in murine and human cells. J. Immunol. 175, 6022–6031 (2005).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 (2011).
Martinez, F.O., Gordon, S., Locati, M. & Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 (2006).
Lacey, D.C. et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J. Immunol. 188, 5752–5765 (2012).
Richez, C. et al. Role for interferon regulatory factors in autoimmunity. Joint Bone Spine 77, 525–531 (2010).
Hedl, M. & Abraham, C. IRF5 risk polymorphisms contribute to interindividual variance in pattern recognition receptor-mediated cytokine secretion in human monocyte-derived cells. J. Immunol. 188 (2012).
Xu, H. et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol. 13, 642–650 (2012).
Carta, S. et al. The rate of interleukin-1beta secretion in different myeloid cells varies with the extent of redox response to Toll-like receptor triggering. J. Biol. Chem. 286, 27069–27080 (2011).
Qu, Y., Franchi, L., Nunez, G. & Dubyak, G.R. Nonclassical IL-1 beta secretion stimulated by P2 × 7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 179, 1913–1925 (2007).
Pelegrin, P., Barroso-Gutierrez, C. & Surprenant, A. P2 × 7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J. Immunol. 180, 7147–7157 (2008).
Tang, D., Kang, R., Coyne, C.B., Zeh, H.J. & Lotze, M.T. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158–175 (2012).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Piccini, A. et al. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 105, 8067–8072 (2008).
Netea, M.G. et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335 (2009).
Seshadri, S., Duncan, M.D., Hart, J.M., Gavrilin, M.A. & Wewers, M.D. Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1beta processing and release. J. Immunol. 179, 1274–1281 (2007).
Ward, J.R. et al. Temporal interleukin-1beta secretion from primary human peripheral blood monocytes by P2 × 7-independent and P2 × 7-dependent mechanisms. J. Biol. Chem. 285, 23147–23158 (2011).
Cheung, R., Ravyn, V., Wang, L., Ptasznik, A. & Collman, R.G. Signaling mechanism of HIV-1 gp120 and virion-induced IL-1beta release in primary human macrophages. J. Immunol. 180, 6675–6684 (2008).
Chen, H., Cowan, M.J., Hasday, J.D., Vogel, S.N. & Medvedev, A.E. Tobacco smoking nhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. J. Immunol. 179, 6097–6106 (2007).
Novikov, A. et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J. Immunol. 187, 2540–2547 (2011).
Vogt, G. & Nathan, C. In vitro differentiation of human macrophages with enhanced antimycobacterial activity. J. Clin. Invest. 121, 3889–3901 (2012).
Chapman, S.J. & Hill, A.V. Human genetic susceptibility to infectious disease. Nat Rev Genet 13, 175–188 (2012).
Le Moine, O. et al. Role of defective monocyte interleukin-10 release in tumor necrosis factor-alpha overproduction in alcoholics cirrhosis. Hepatology 22, 1436–1439 (1995).
Gleeson, M. et al. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 11, 607–615 (2011).
Yang, L. et al. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 137, 588–597 (2009).
Barnich, N. et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Invest. 117, 1566–1574 (2007).
Garrett, W.S., Gordon, J.I. & Glimcher, L.H. Homeostasis and inflammation in the intestine. Cell 140, 859–870 (2010).
Ley, R.E., Turnbaugh, P.J., Klein, S. & Gordon, J.I. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Jacob, C.O. et al. Heritable major histocompatibility complex class II-associated differences in production of tumor necrosis factor alpha: relevance to genetic predisposition to systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 87, 1233–1237 (1990).
Panda, A. et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol. 184, 2518–2527 (2010).
Wurfel, M.M. et al. Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am. J. Respir. Crit. Care Med. 178, 710–720 (2008).
Wurfel, M.M. et al. Identification of high and low responders to lipopolysaccharide in normal subjects: an unbiased approach to identify modulators of innate immunity. J. Immunol. 175, 2570–2578 (2005).
Vamvakopoulos, J., Green, C. & Metcalfe, S. Genetic control of IL-1beta bioactivity through differential regulation of the IL-1 receptor antagonist. Eur. J. Immunol. 32, 2988–2996 (2002).
von Bernuth, H. et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321, 691–696 (2008).
Sancho-Shimizu, V. et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Invest. 121, 4889–4902 (2011).
Arnott, I.D. et al. NOD2/CARD15, TLR4 and CD14 mutations in Scottish and Irish Crohn’s disease patients: evidence for genetic heterogeneity within Europe? Genes Immun. 5, 417–425 (2004).
Bonen, D.K. et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 124, 140–146 (2003).
Ferwerda, B. et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc. Natl. Acad. Sci. USA 104, 16645–16650 (2007).
Schadt, E.E. Molecular networks as sensors and drivers of common human diseases. Nature 461, 218–223 (2009).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Garrett, W.S. et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45 (2007).
Maslowski, K.M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009).
Shultz, L.D., Brehm, M.A., Garcia-Martinez, J.V. & Greiner, D.L. Humanized mice for immune system investigation: progress, promise and challenges. Nat. Rev. Immunol. 12, 786–798 (2012).
Karban, A.S. et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum. Mol. Genet. 13, 35–45 (2004).
Garlanda, C., Anders, H.J. & Mantovani, A. TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 30, 439–446 (2009).
Chen, X., Zhao, Y., Wu, X. & Qian, G. Enhanced expression of single immunoglobulin IL-1 receptor-related molecule ameliorates LPS-induced acute lung injury in mice. Shock 35, 198–204 (2011).
Wald, D. et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 4, 920–927 (2003).
Garlanda, C. et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA 101, 3522–3526 (2004).
Lech, M. et al. Tir8/Sigirr prevents murine lupus by suppressing the immunostimulatory effects of lupus autoantigens. J. Exp. Med. 205, 1879–1888 (2008).
Horne, D.J. et al. Common polymorphisms in the PKP3-SIGIRR-TMEM16J gene region are associated with susceptibility to tuberculosis. J. Infect. Dis. 205, 586–594 (2012).
Adib-Conquy, M. et al. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit. Care Med. 34, 2377–2385 (2006).
Mitsuzawa, H. et al. Recombinant soluble forms of extracellular TLR4 domain and MD-2 inhibit lipopolysaccharide binding on cell surface and dampen lipopolysaccharide-induced pulmonary inflammation in mice. J. Immunol. 177, 8133–8139 (2006).
Raby, A.C. et al. Soluble TLR2 reduces inflammation without compromising bacterial clearance by disrupting TLR2 triggering. J. Immunol. 183, 506–517 (2009).
Gazouli, M. et al. Association between polymorphisms in the Toll-like receptor 4, CD14, and CARD15/NOD2 and inflammatory bowel disease in the Greek population. World J. Gastroenterol. 11, 681–685 (2005).
Lakatos, P.L. et al. Serum lipopolysaccharide-binding protein and soluble CD14 are markers of disease activity in patients with Crohn’s disease. Inflamm. Bowel Dis. 17, 767–777 (2012).
Spencer, S.D. et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J. Exp. Med. 187, 571–578 (1998).
Mauro, C. et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem 281, 18482–18488 (2006).
Gao, L. et al. ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibit antiviral signaling. J. Biol. Chem. 286, 36592–36602 (2011).
Zhou, J. et al. A20-binding inhibitor of NF-kappaB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein beta activation and protects from inflammatory disease. Proc. Natl. Acad. Sci. USA 108, E998–1006 (2011).
Nanda, S.K. et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J. Exp. Med. 208, 1215–1228 (2011).
El Bakkouri, K., Wullaert, A., Haegman, M., Heyninck, K. & Beyaert, R. Adenoviral gene transfer of the NF-kappa B inhibitory protein ABIN-1 decreases allergic airway inflammation in a murine asthma model. J. Biol. Chem. 280, 17938–17944 (2005).
Wullaert, A. et al. Adenoviral gene transfer of ABIN-1 protects mice from TNF/galactosamine-induced acute liver failure and lethality. Hepatology 42, 381–389 (2005).
Gateva, V. et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233 (2009).
Han, J.W. et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 41, 1234–1237 (2009).
Wullaert, A. et al. LIND/ABIN-3 is a novel lipopolysaccharide-inducible inhibitor of NF-kappaB activation. J. Biol. Chem. 282, 81–90 (2007).
Mashima, R. et al. FLN29, a novel interferon- and LPS-inducible gene acting as a negative regulator of toll-like receptor signaling. J. Biol. Chem. 280, 41289–41297 (2005).
Sanada, T. et al. FLN29 deficiency reveals its negative regulatory role in the Toll-like receptor (TLR) and retinoic acid-inducible gene I (RIG-I)-like helicase signaling pathway. J. Biol. Chem. 283, 33858–33864 (2008).
Chi, H. et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. USA 103, 2274–2279 (2006).
Matta, R. et al. Knockout of Mkp-1 exacerbates colitis in Il-10-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G1322–G1335 (2012).
Frazier, W.J. et al. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J. Immunol. 183, 7411–7419 (2009).
Bhavsar, P. et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax 63, 784–790 (2008).
Rastogi, R. et al. Dysregulation of p38 and MKP-1 in response to NOD1/TLR4 stimulation in sarcoid bronchoalveolar cells. Am. J. Respir. Crit. Care Med. 183, 500–510 (2011).
Burns, K. et al. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 197, 263–268 (2003).
Zaki, M.H. et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 (2011).
Hubbard, L.L. et al. A role for IL-1 receptor-associated kinase-M in prostaglandin E2-induced immunosuppression post-bone marrow transplantation. J. Immunol. 184, 6299–6308 (2010).
Aronoff, D.M., Canetti, C. & Peters-Golden, M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 173, 559–565 (2004).
Serezani, C.H. et al. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 37, 562–570 (2007).
Kabashima, K. et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J. Clin. Invest. 109, 883–893 (2002).
Morteau, O. et al. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J. Clin. Invest. 105, 469–478 (2000).
Chong, M.M., Metcalf, D., Jamieson, E., Alexander, W.S. & Kay, T.W. Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood 106, 1668–1675 (2005).
Mottok, A., Renne, C., Willenbrock, K., Hansmann, M.L. & Brauninger, A. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood 110, 3387–3390 (2007).
Berlato, C. et al. Involvement of suppressor of cytokine signaling-3 as a mediator of the inhibitory effects of IL-10 on lipopolysaccharide-induced macrophage activation. J. Immunol. 168, 6404–6411 (2002).
Croker, B.A. et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 4, 540–545 (2003).
Roberts, A.W. et al. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. USA 98, 9324–9329 (2001).
Yasukawa, H. et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 4, 551–556 (2003).
Schreiber, S. et al. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut 51, 379–385 (2002).
Bulut, Y., Faure, E., Thomas, L., Equils, O. & Arditi, M. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J. Immunol. 167, 987–994 (2001).
Zhang, G. & Ghosh, S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J. Biol. Chem. 277, 7059–7065 (2002).
Burns, K. et al. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell. Biol. 2, 346–351 (2000).
Didierlaurent, A. et al. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol. Cell Biol. 26, 735–742 (2006).
Schimming, T.T. et al. Association of toll-interacting protein gene polymorphisms with atopic dermatitis. BMC Dermatol. 7, 3 (2007).
Shah, J.A. et al. Human TOLLIP regulates TLR2 and TLR4 signaling and its polymorphisms are associated with susceptibility to tuberculosis. J. Immunol. 189, 1737–1746 (2012).
Sosic, D., Richardson, J.A., Yu, K., Ornitz, D.M. & Olson, E.N. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 112, 169–180 (2003).
Sharabi, A.B. et al. Twist-2 controls myeloid lineage development and function. PLoS Biol. 6, e316 (2008).
Chen, Z.F. & Behringer, R.R. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 9, 686–699 (1995).
Niesner, U. et al. Autoregulation of Th1-mediated inflammation by twist1. J. Exp. Med. 205, 1889–1901 (2008).
Acknowledgements
We gratefully acknowledge Judy Cho for critical reading of the manuscript. This work was supported by R01DK077905, DK-P30-34989, and U19-AI082713 (CA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Hedl, M., Abraham, C. Negative regulation of human mononuclear phagocyte function. Mucosal Immunol 6, 205–223 (2013). https://doi.org/10.1038/mi.2012.139
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2012.139
This article is cited by
-
Diversity and functions of intestinal mononuclear phagocytes
Mucosal Immunology (2017)
