
Abstract
Myocardial fibrosis is characterized by significant extracellular matrix (ECM) deposition. The specific cellular mediators that contribute to the development of fibrosis are not well understood. Using a model of fibrosis with Angiotensin II (AngII) infusion, our aim was to characterize the cellular elements involved in the development of myocardial fibrosis. Male C57Bl/6 and Tie2-GFP mice were given AngII (2.0 mg/kg/min) or saline (control) via mini osmotic pumps for up to 7 days. Hearts were harvested, weighed and processed for analysis. Cellular infiltration and collagen deposition were quantified. Immunostaining was performed for specific markers of leukocytes (CD45, CD11b), myofibroblasts (SMA), endothelial cells (vWF) and hematopoietic progenitor cells (CD133). Bone marrow (BM) origin of infiltrating cells was assessed using GFP+ chimeric animals. Relative qRT–PCR was performed for pro-fibrotic cytokines (transforming growth factor (TGF)-β1, CTGF) as well as the chemokine stromal-derived factor (SDF)-1α. Myocardial-infiltrating cells were grown in vitro. AngII exposure resulted in multifocal myocardial cellular infiltration, which preceded extensive ECM deposition. A limited number of myocardial-infiltrating cells were positive for leukocyte markers but were significantly positive for myofibroblast (SMA) and endothelial cell (vWF) markers. However, using Tie2-GFP mice, where endothelial cells are GFP+, myocardial-infiltrating cells were not GFP+. Transcript levels for SDF-1α were significantly elevated at 1 day of AngII exposure suggesting that hematopoietic progenitor cells may be recruited. This was confirmed by positive CD133 staining of infiltrating cells and evident GFP+ cellular infiltration when exposing GFP+ BM chimeras to AngII. Furthermore, a significant number of CD133+/SMA+ cells were grown in vitro from the myocardium of AngII-exposed animals (P<0.01). Myocardial ECM deposition is preceded by the infiltration of the myocardium with hematopoietic cells that express mesenchymal markers. These data suggest that mesenchymal progenitor cells are recruited, and may have a primary role, in the initiation of myocardial fibrosis.
Similar content being viewed by others


Main
Myocardial fibrosis is a common pathological feature seen in many patients with heart disease and is hypothesized to be the final common pathway that ultimately results in irreversible organ failure.1 Pathologically, fibrosis is characterized by the accumulation of extracellular matrix (ECM) proteins within the myocardium. While initially ECM deposition can be beneficial and occur in response to injury, the repair response can become exacerbated.1 The lack of resolution of this ECM deposition in tissue is the hallmark of fibrosis.2 The accumulation of ECM proteins within the myocardium enhances cardiac stiffness and can eventually impair cardiac function leading to the development of heart failure.1 The exact mechanisms responsible for myocardial fibrosis remain to be clearly defined, despite intense study.
Recent evidence has implicated Angiotensin II (AngII) in the progression of myocardial fibrosis. AngII has been suggested to be a potent pro-fibrotic molecule.1 Increased serum levels of AngII are seen in patients with cardiovascular diseases that are associated with myocardial fibrosis, including atherosclerosis, hypertension, cardiac hypertrophy and heart failure.1, 3, 4 Clinical trials looking at the inhibition of the renin-angiotensin system, and more specifically AngII receptor blockers (ARB), demonstrate significant benefit in the prevention of cardiovascular events, including the cardiac remodeling seen with myocardial fibrosis.5, 6 Moreover, AngII given exogenously to rodents has been shown to result in cellular changes within the myocardium, hypertrophy and eventual fibrosis, similar to that seen in humans.5, 7, 8, 9 Taken together, this evidence strongly supports a role for AngII in the development of myocardial fibrosis. The direct mechanisms responsible, and the effector cells involved, have yet to be fully characterized.
Rapid myocardial cellular infiltration is evident in animals infused with AngII, which suggests that the cellular component has an effector function in the development of myocardial fibrosis.10 It is unclear, however, what cell types participate in this infiltration. The cellular accumulation has been primarily described as mononuclear in appearance, lacking polymorphonuclear cells. This has led some investigators to suggest that these cells are of the monocyte/macrophage lineage.7, 8, 11, 12, 13 One should note that cells that stain positive for monocyte/macrophage markers do not account for all the infiltrating cells.7, 8, 11 This observation suggests a role for a yet to be described cell type.
The predominant characteristic of fibrosis, ECM deposition, is generally thought to result from the activities of fibroblasts and myofibroblasts rather than macrophages. Cardiac fibroblasts are activated in response to hypertrophic stimuli, including AngII.14, 15, 16, 17 Furthermore, fibroblasts proliferate and increase the production of ECM proteins in response to AngII exposure in vitro. However, the in vivo data available to support this response to AngII is limited.1, 14, 15, 16, 17 Finally, if myofibroblasts make up a component of the cells that accumulate in the myocardium after AngII exposure, their origin remains to be determined. Myofibroblasts can arise from the activation and proliferation of resident fibroblasts or from several different sources including endothelial–mesenchymal transitions and blood borne mesenchymal progenitor cells (referred to as fibrocytes).2, 18, 19, 20, 21
The data presented in this study characterize early cellular migration to the myocardium in response to AngII. We identify a novel population of hematopoietic progenitor cells with mesenchymal properties as an infiltrating cellular population involved in AngII-mediated myocardial fibrosis and as such, suggest alternative mechanisms in the initiation of fibrosis.
MATERIALS AND METHODS
Animals
All work was approved by Dalhousie University's University Committee on Laboratory Animals. Male C57BL/6, enhanced green fluorescent progein (eGFP) and Tie2-GFP mice ranging from 7 to 8 weeks of age were purchased from Jackson Laboratory (Bar Harbour, ME, USA) and were housed within the Medical Sciences Animal Care Facility at Dalhousie University. Mice were provided food and water ad libitum for 1 week prior experimentation.
AngII Infusion
Animals were anesthetized with isoflurane (Baxter Healthcare, New Providence, NJ, USA) in oxygen delivered by a Fortec vaporizer. When surgical levels of anesthesia were reached, a 1–2 cm mid-scapular skin incision was made and a mini osmotic pump (Alzet, Palo Alto, CA, USA) was inserted subcutaneously. The incision was closed using 7 mm wound clips. Animals were randomly assigned to receive AngII (2.0 μg/kg/min; Sigma Aldrich, Oakville, ON, USA), AngII and Losartan (ARB; 6.9 μg/kg/min, Sigma Aldrich) or a vehicle control of saline. The pumps remained in for 1, 3 or 7 days during which the animals were provided with food and water ad libitum and observed for signs of morbidity. Prior to euthanization, blood pressure measurements were taken via the Coda2 non-invasive cuff system (Kent Scientific, Torrington, CT, USA) for a minimum of five consecutive measurements.
GFP+ Bone Marrow Chimera
Generation of GFP bone marrow (BM) chimeric animals were generated as previously described.22 Briefly, BM cells were harvested from 8-week-old eGFP-transgenic mice with a C57Bl/6 background.22 After irradiation with a two doses of 5.5 Gy, 4 h apart, the unfractionated GFP+ BM cells (5 × 106 cells in 150 μl of sterile phosphate-buffered saline) were injected via the tail vein into irradiated C57/Bl6 mice. Engraftment was evaluated 8 weeks later by collecting peripheral blood cells and looking at the frequency of GFP+ cells among peripheral nucleated cells (data not shown).22, 23 Only animals with demonstrated engraftment (>60% of white blood cells were GFP+) were used for the two treatment groups (saline control or AngII infusion as per protocol). Immunofluorescence was used to enhance localize GFP+ cells within the myocardium. Antigens were retrieved on sections of formalin-fixed and paraffin-embedded GFP chimeric tissues and then non-specific protein binding was blocked with 10% normal goat serum. The tissues were then incubated with the rabbit anti-eGFP primary antibody (Abcam, Cambridge, MA, USA) followed by incubation with an alexa488-conjugated goat anti-rabbit. Tissues were thoroughly washed and cover slipped. Fluorescent microscopy was performed; pictures were captured and analyzed in Adobe Photoshop 5.0. Cover slips were removed and the tissues were then stained with a hematoxin and eosin (H&E) stain to assess basic histology of tissues for comparison to GFP positivity. Light microscopy was performed; pictures were captured and analyzed in Adobe Photoshop 5.0.
Tissue Harvest
Hearts from experimental animals were harvested and weighed. Cardiac mass index was measured by calculating the heart:body weight ratio. The hearts were lastly divided along the short axis into three portions including the base, middle and apical sections. The base portion was processed for histological examination while the other two portions were snap frozen immediately for molecular analysis.
Cell Isolation and Culture
Hearts from untreated mice (n=3) or mice infused with AngII for 3 days (n=3) were harvested under sterile conditions and used for cell isolation. Briefly, hearts were initially mechanically minced and then enzymatically digested in a Collagenase solution (50 μg/ml Collagenase II, Cedar Lane, Burlington, ON, USA) in Roswell Park Memorial Institute 1640 (RPMI) media (Gibco, Life Technologies, Burlington, ON, USA) at 37°C with agitation for 45 min. The cell isolates were washed twice with complete RPMI (10% heat-inactivated fetal calf serum, 2 mM L-glutamate, 100 μg/ml streptomycin and 100 U/ml Penicillin) and then plated on T-25 flasks coated with 0.1% gelatin. Cellular isolates were incubated at 37°C with 5% CO2 for 3 days, after which all non-adherent cellular debris was removed and fresh media was supplied. In all groups, digital images were taken using an inverted microscope for cell counting. Areas of cell growth were randomly selected (6/flask containing cells isolated from one heart) for cell counts. Adherent cells from naive hearts were then serum starved for 24 h and then exposed to AngII (100 nM) in serum-free RPMI for 2 days. Digital images were again taken to assess cell growth.
For immunofluorescence, cultured myocardial cells initially grown on 0.1% gelatin-coated cover slips were fixed with 4% paraformaldehyde for 30 min. Non-specific protein binding was blocked with a Mouse on Mouse Block kit (Vector, Burlington, CA, USA) and normal goat serum. The cells were incubated with the primary antibodies of rabbit anti-CD133 (Abcam) and mouse anti-α-smooth muscle actin (SMA, Sigma Aldrich). This was followed by incubation with an Alexa488-conjugated goat anti-mouse secondary and an Alexa555-conjugated goat anti-rabbit secondary. Cells were thoroughly washed and mounted on slides. Fluorescent microscopy was performed, and digital images were acquired and analyzed in Adobe Photoshop 5.0. Furthermore, differential interference contrast microscopy was performed and digital images were acquired.
Histological Analysis
Hearts were processed for histological assay by: (a) fixing with 10% formalin for 24 h; (b) protecting with sucrose/OCT followed by snap freezing or (c) fixing with 4% paraformaldehyde for 24 h. Formalin-fixed tissues were paraffin embedded and were serially sectioned on a microtome (5 μm). Frozen and paraformadehyde-fixed tissues were OCT embedded and serially sectioned on a cryostat (8 μm). Basic myocardial histology and cellular infiltration were examined using heart cross-sections stained with H&E. Cellular changes were quantified by using an overlaid 500 × 500 pixel grid over heart sections, which were captured at × 5 magnification. Grids that contained any cellular infiltration or areas of excess ECM deposition were tallied as well as total grids encompassed by the heart. The percent of grids affected were then calculated. Immunohistochemistry for SMA (Sigma Aldrich), von Willebrand Factor (vWF; Chemicon, Temecula, CA, USA), KI-67 (DakoCytomation, Mississauga, ON, USA) and CD133 (Abcam) was performed on paraffin-embedded tissues which required deparaffinization and antigen retrieval prior to staining. Immunohistochemistry was performed for CD45 (BD Biosciences, San Jose, CA, USA) and CD11b (AbD Serotec, Raleigh, NC, USA) on frozen tissue, which did not require antigen retrieval prior to staining. Briefly, endogenous peroxidases were quenched with 3% hydrogen peroxide; endogenous biotin was blocked (DAKO Biotin Blocking System, DakoCytomation); and non-specific staining was blocked with normal goat serum or normal horse serum. Sections were incubated with the individual primary antibodies and followed by a specific biotin-conjugated secondary antibody. The antibody complexes were then conjugated to an Avidin–biotin complex (Vecstastain ABC kit; Vector) and developed using 3,3′diaminobenzidine as the chromogen (DAB, DakoCytomation). Light microscopy was performed; pictures were captured and analyzed in Adobe Photoshop 5.0. Immunofluorescent staining was also performed for vWF on paraformaldehyde-fixed tissues. Briefly, the tissues were incubated with normal goat serum to block non-specific binding. The slides were incubated with the primary antibody followed by a Cy3-conjugated secondary. Fluorescent microscopy was performed; pictures were captured and analyzed in Adobe Photoshop 5.0.
Collagen Deposition
Collagen detection was accomplished using Sirius red and fast green stains, and quantified using a modified technique originally described by Underwood et al.24 Image analysis software (IPTK 5.0, Reindeer Graphics, Asheville, NC, USA) was used to quantify collagen in the heart sections. Briefly, slides were examined under a microscope with the × 10 objective and areas of collagen deposition were captured with a digital camera. The image analysis software was used to quantify the amount of tissue positive for Sirius red. Sections from individual animals were averaged and experimental groups were compared to control animals.
Relative Quantitative Polymerase Chain Reaction (qRT–PCR)
Using the TRIzol reagent (Gibco), RNA was isolated from the snap frozen myocardium as per the manufacturer's protocol. First strand cDNA was synthesized from 1 μg of total RNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA).
Relative quantitative RT–PCR (qRT–PCR) was completed using 12.5 ng of input cDNA with 0.5 μM of each of the forward and reverse primers and 1 × iQ SYBR Green Supermix (Bio-Rad) was subjected to qRT–PCR using iQ Multicolour Real-Time PCR Detection System thermocycler (Bio-Rad). Standard curves, for efficiency, and no-template control samples were run along with the samples during thermocycling. A melting curve was performed after thermocycling was complete to ensure target specificity. The primers were designed against the mRNA sequence of connective tissue growth factor (CTGF) (forward: 5′-TCAACCTCAGACACTGGTTTCG-3′; reverse: 5′-TAGAGCAGGTCTGTCTGCAAGC-3′), stromal-derived factor (SDF)-1α (forward: 5′-GTAGAATGGAGCCAGACCATCC-3′; reverse: 5′-ATTCGATCAGAGCCCATAGAGC-3′), transforming growth factor (TGF)-β1 (forward: 5′-GACTATCCACCTGCAAGACTATCG-3′; reverse: 5′-GACTATCCACCTGCAAGACTATCG-3′) and 18s ribosomal RNA (forward: 5′-TCAACTTTCGATGGTAGTCGCCGT-3′; reverse: 5′-TCCTTGGATGTGGTAGCCGTTTCT-3′). Expression was normalization to the 18s ribosomal gene using the Pfaffl method.
Statistical Analysis
One-way ANOVA tests were completed on all quantitative data using the Dunnet post-test to compare the experimental groups to the saline control. Our level of significance was set as P≤0.05. All statistical calculations were computed using GraphPad Prism 4 software.
RESULTS
Effect of AngII on Animals
Animals were assigned to three experimental groups and infused with: (a) AngII (2.0 μg/kg/min, n=35); (b) AngII with Losartan (ARB, 6.9 μg/kg/min, n=4) or (c) saline (control, n=17). Myocardial hypertrophy was measured via a cardiac mass index. AngII exposure resulted in a significant increase in cardiac mass index at 7 days (P<0.01) compared to control animals (Figure 1). Infusion of an ARB abolished the hypertrophic effect of AngII at 7 days (Figure 1) confirming that AngII effects are dependent on the interaction with its receptor (AT1R). AngII-exposed animals showed a significant increase in their systolic, diastolic and mean arterial pressures after 7 days of exposure when compared to control animals (P<0.01; Figure 1).

The effects of AngII on animals; AngII exposure induced an increase in the cardiac mass index (heart weight:body weight ratio) at 7 days (a); hemodynamic measurements were taken via tail cuff and AngII exposure at 7 days increased systolic, diastolic and mean arterial pressure (b). **P<0.01.
Myocardial Collagen Deposition
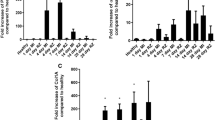
Hearts were harvested after 1, 3 and 7 days and the degree of myocardial collagen deposition was quantified in heart sections stained with Sirius red. AngII exposure resulted in multifocal areas of extensive collagen deposition after 3 and 7 days when compared to control animals (Figure 2). Quantification of collagen accumulation in AngII-exposed animals confirmed these findings at 3 and 7 days post-AngII infusion compared to background collagen levels seen in control animals (P<0.01; Figure 2). Co-treatment of mice with AngII and an ARB resulted in baseline levels of collagen deposition suggesting that the fibrotic effects of AngII require AT1R signaling. The transcript levels of the pro-fibrotic factor TGF-β1, analyzed by qRT–PCR, was significantly elevated above saline levels after 1 and 3 days of exposure (four- and fivefold increase, respectively, P<0.01; Figure 2). Similarly, the transcript levels of the pro-fibrotic factor CTGF downstream of TGF-β1, analyzed by qRT–PCR, was also significantly elevated above saline levels after 1 and 3 days of exposure (19- and 13-fold increase, respectively, P<0.01; Figure 2), but were at baseline levels in animals receiving AngII and an ARB (Figure 2). The significant increase in both TGF-β1 and CTGF transcript levels at 1 day appears to temporally precede the increase in collagen deposition seen at 3 and 7 days.

Collagen deposition: to evaluate collagen deposition heart sections were stained with Sirius red and fast green. Representative sections are shown from control animals (a), 7 days AngII-exposed animals (b) and 7 days AngII-exposed animals+ARB (c). Collagen in the heart sections was quantified using image analysis software. AngII exposure resulted in significant collagen deposition at 3 and 7 days and appeared dependent on AngII receptor mechanism (d). Using qRT–PCR, we measured myocardial expression of TGF-β1 (e) and CTGF (f) mRNA transcript levels relative to the 18s ribosomal gene expression. Relative mRNA transcript levels for both TGF-β1 and CTGF were significantly increased in AngII infused animals at 1 and 3 days, **P<0.01. Images were captured at × 25.
Cellular Infiltration
Hearts stained with H&E suggest that AngII exposure resulted in progressive myocardial changes over the 7-day period. Multifocal areas of cellular infiltration and myocyte loss were evident, particularly at 3 and 7 days, in the myocardium of AngII-exposed animals (Figure 3). We have adapted a grid-scoring method to quantify cellular infiltration within AngII-exposed myocardiums confirming that significant myocardial cellular infiltration is evident as early as 1 day exposure and progressively increased at 3 and 7 days (P<0.01 at 1, 3 and 7 days; Figure 3). In contrast, hearts from saline control animals and animals co-treated with AngII and an ARB exhibited normal myocardial histology with no significant cellular infiltration (Figure 3).

Heart morphology: representative sections of hearts stained with H&E from animals: saline control (a) AngII for 7 days (b) and 7 days AngII-exposed animals+ARB (c). AngII infused animals resulted in areas of marked mononuclear cell accumulation, which preceded cardiomyocyte loss and replacement by extracellular matrix. The percent area of the myocardium affected by foci of myocardial cellular infiltration and ECM deposition was quantified to confirm visual assessments (d), **P<0.01. Images were captured at × 25.
Cells seen infiltrating the myocardium were mononuclear in appearance with no evident polymorphonuclear cells. Immunohistochemistry was used to identify these cells using cell-specific markers. At 7 days, <20% of the infiltrating cells were positive for a leukocyte-specific marker (CD45; Figure 4), which also corresponded with staining patterns for macrophage-specific markers (CD11b; Figure 4) and negative peroxidase stain (data not shown). In contrast, immunohistochemistry for myofibroblasts/smooth muscle cells (SMA) and endothelial cells (vWF) showed a large number of the infiltrating cells stain positively in areas of ECM deposition (Figure 4).


Immunocytochemistry: immunostaining for a pan-leukocyte marker (CD45, a, b), monocyte/macrophage marker (CD11b, c, d), myofibroblasts (SMA, e, f), and endothelial cell associated marker (vWF, g, h). Representative sections are shown from control animals (a, c, e, g), and 7 days AngII-exposed animals (b, d, f, h). AngII exposure is associated with myocardial infiltration with some CD45+ and CD11b+ cells but marked SMA+ and vWF+ cells. Images were captured at × 63.
The evident vWF staining is indicative of endothelial cell accumulation suggestive of neo-angiogenesis. To confirm the presence of endothelial cells, we used a transgenic mouse (Tie2-GFP) that expresses GFP under the endothelial-specific promoter Tie2. In the myocardium of Tie2-GFP mice exposed to AngII, few GFP-positive cells were visible within areas of myocardial cellular infiltration and little co-localization with vWF-positive staining was evident (Figure 5). These data suggest that the infiltrating cells are not differentiated endothelial cells.

Endothelial cell localization: fluorescence of natural green fluorescence protein (GFP) expression in a Tie2-GFP mouse (a, d), immunofluorescence of vWF staining with a red Cy3 secondary (b, e) and the combined image of both the GFP and vWF fluorescence with yellow coloration indicating overlapped expression (c, f). Representative images from control animals (a–c) and 7 days AngII animals (d–f) are shown. Images were captured at × 63.
Origin of Myocardial-Infiltrating Cells
Based on KI-67 staining, infiltrating cells appear to undergo significant proliferation suggesting that they may be undifferentiated cells with a high proliferative index (Figure 6). The transcript levels for a well-known chemokine for hematopoietic progenitor cells, SDF-1α, was significantly increased within 1 day of exposure to AngII and remained elevated over the 7-day exposure period (Figure 7). We used immunohistochemistry for a hematopoietic progenitor cell marker, CD133, to identify and localize any hematopoietic progenitor cells in vivo. A large population of CD133+ cells was evident within the areas of myocardial infiltration in AngII-exposed myocardiums at 3 and 7 days of exposure, which corresponded to the expression of SMA+ marker (Figure 7). The BM origin of these myocardial-infiltrating cells was then confirmed using GFP+ chimera mice. GFP+ BM chimera (n=5) mice were exposed to AngII or saline infusion and heart tissue examined at 3 days. Virtually all infiltrated cells were positive for GFP when compared to saline control (Figure 8).

Cellular proliferation: representative images of immunocytochemistry for the proliferation marker KI-67 in control animals (a) and 7 days AngII animals (b). Images were captured at × 63.

Hematopoietic progenitor cells: immunostaining for the hematopoietic progenitor cell marker CD133. Representative sections are shown from control animals (a), 3 days AngII-exposed animals (b) and 7 days AngII-exposed animals (c). AngII exposure is associated with myocardial infiltration by a CD133+ population increasing from 3 to 7 days. Using qRT–PCR, transcript level of SDF-1α (d) was measured and normalized to 18s ribosomal gene expression. Relative transcript levels of SDF-1α was significantly increased in AngII infused animals. **P<0.01. Images were captured at × 63.

GFP chimera experiments: representative sections of heart tissue taken from GFP+ BM chimeric animals that were assigned at random: saline control treatment for 3 days (a, b) or AngII for 3 days (c, d). AngII infusion resulted in areas of marked cellular accumulation seen in H&E (c) when compared to control (a). Furthermore, all areas of cellular infiltration in AngII animals were strongly positive for GFP+ immunofluorescence (d) when compared to saline control (b), suggesting that virtually all infiltrating cells are of BM origin. Images were captured at × 25.
Isolation of CD133+/SMA+and Growth In Vitro
Using whole heart homogenate, we were able to grow primary cultures of adherent cells in an effort to characterize the origin of myocardial-infiltrating cells seen in animals exposed to AngII. Myocardial cell isolates were taken from both AngII-exposed and untreated animals. After 3 days in culture, a distinct adherent cell population with spindle shape morphology was evident in flasks containing isolates from AngII-treated myocardiums (Figure 9). This was in contrast to very few adherent cells seen in cultures of untreated heart homogenates (Figure 9). When quantified, there were significantly more adherent cells in cultures from AngII treated as compared to untreated hearts (60 and 8 cells/HPV, respectively, P<0.01; Figure 9). Dual labeling for CD133 and SMA using immunofluorescent labels was used to further characterize the cultured cell. Cells from untreated hearts were single-positive CD133−SMA+ whereas 81±3% of cells from an AngII-exposed heart appeared to be double-positive CD133+SMA+ (Figure 9). In an additional set of experiments, cells cultured from untreated hearts were treated with AngII (100 nM) for 2 days but failed to show a significant expansion in the number of CD133+ cells, further supporting that their origin is not from resident cells (Figure 9).


Isolated cells: cells were isolated out of the myocardium of untreated and 3 days AngII-exposed animals. Photomicrograph were taken of adherent cell populations obtained from untreated (a, c, e, g) and AngII-exposed (b, d, f, h) myocardium after 3 days in culture at × 40 magnification. Representative pictures illustrate the increase in adherent cells in cultures from AngII-exposed myocardiums (g, h). Cell counting (6 high power field (HPV) × 10/animal) was used to quantify the degree of cell growth by adherent cells between untreated naive cells, untreated cells exposed to AngII (100 nM) and cells from AngII-exposed myocardium (i). Dual labeling immunofluorescence was used to demonstrate co-localization of SMA (green; a, b), CD133 (red: c, d), and overlaid to show co-localization of markers (yellow) (e, f) captured at × 40 magnification. **P<0.01.
DISCUSSION
We have shown that AngII exposure results in significant ECM deposition that is characteristic of myocardial fibrosis. This collagen deposition was correlated with an increase in key pro-fibrotic factors, TGF-β1 and its downstream mediator CTGF, shortly after AngII infusion. We have provided evidence that early cellular infiltration into the myocardium precedes significant ECM deposition, potentially providing pro-fibrotic stimuli to initiate fibrosis. Despite the fact that the AngII infusion model is a well-established model for hypertrophy, fibrosis and hypertension, there has been little work done to characterize the cellular component of the response. The current paradigm asserts that fibrosis is the response to an initial stimulus that induces local inflammation and the recruitment of inflammatory cells. These cells then stimulate a fibrotic response. This theory has been supported by the identification of mononuclear cells, positive for macrophage markers, in the infiltrating cell population.7, 8, 11, 12, 13 Cell phenotypes, and marker expression, are dynamic and cells can alter their molecular expression to respond appropriately to stimuli in their environmental milieu. There is a circulating monocyte/macrophage precursor population that expresses specific markers for this cell type but are a phenotypically heterogeneous population.25, 26 Furthermore, precursors from the monocyte/macrophage lineage have been found to differentiate into non-immune cell types when presented with the appropriate stimuli.26, 27, 28 It is plausible that macrophage marker expression in tissue could have a temporal relationship to the degree of cell differentiation. We found very few cells expressing leukocyte or macrophage markers within the myocardium of mice early after AngII infusion which has been supported by others.29 These data suggest that fully differentiated leukocytes, specifically macrophages, may not represent the primary infiltrating cell. These data cast doubt on the prevailing paradigm that acute inflammation is the primary mechanism promoting fibrosis. However, there is an important distinction to be made between the present study, which uses a model of AngII-mediated fibrosis, compared to ischemic models that have been associated with fibrosis. In models of ischemia, in which there has been myocardial injury or infarction, inflammation has been shown to have a key function in healing and likely participates in the development of fibrosis but its contribution to date has been poorly defined.30, 31, 32 The complex interactions between ischemia and the development of fibrosis were not explored in this study.
We have shown that AngII exposure results in significant positive staining for markers of mesenchymal cells, including SMA and vWF, by early myocardial-infiltrating cells. SMA is a structural protein largely found in smooth muscle cells and myofibroblasts.33 The diffuse staining pattern of SMA would correspond with a myofibroblast-like cell population, which are believed to be the primary cell responsible for ECM deposition.33 vWF, however, is a glycoprotein that is largely concentrated on the subendothelial matrix and as such, is often used as a histological marker for endothelial cells.34 Extensive staining with vWF is indicative of endothelial cell accumulation and/or neo-angiogenesis. However, our findings using Tie2-GFP mice, in which endothelial cells express GFP, suggest that areas of myocardial cellular infiltration do not represent terminally differentiated endothelial cells.
We also found that a significant proportion of the infiltrating cells within the myocardium were positive for CD133. CD133 is known as a marker of hematopoietic stem cells found largely in the BM and circulation.35 The presence of CD133-positive cells within the myocardium of AngII-exposed animals suggests a role for undifferentiated hematopoietic progenitor cells in the fibrotic response. The hematopoetic (BM) origin of infiltrating cells was confirmed using BM chimera in which virtually all infiltrating cells were GFP+. This hypothesis is supported by the fact that SDF-1α, a common chemokine for hematopoietic progenitor cells,36 was significantly upregulated within the myocardium of AngII-exposed animals as early as day 1. This provides a potential mechanism to allow recruitment of circulating progenitor cells out of the blood into the tissue. Furthermore, we have shown that a large proportion of cells accumulating within AngII-exposed myocardium were positive for a proliferation marker, KI-67, suggesting a high proliferative index. Terminally differentiated cells, including myocytes and leukocytes, typically do not undergo proliferation. Collectively, our data suggest that AngII results in the early recruitment of a hematopoietic progenitor cell into the myocardium.
The co-localization of a mesenchymal marker (SMA) and a hematopoietic progenitor marker (CD133) within areas of myocardial cellular infiltration suggests that these cells could represent fibroblast precursor cells, sometimes referred to as ‘fibrocytes’. Fibrocytes are thought by some to be derived from an early monocyte precursor lineage.27 They are characterized by the co-expression of a hematopoietic marker (CD133/CD34), a leukocyte marker (CD45/CD11b) and a mesenchymal marker (SMA/Coll1).37 Expression levels of these markers vary depending on the level of differentiation from early progenitor markers to late myofibroblast markers.37 However, the co-localization of a hematopoietic marker (CD133, CD34) and a mesenchymal marker (SMA, Coll1), as we demonstrated in primary isolated cells in vitro, is sufficient to confirm a fibrocyte phenotype.38 Our in vitro data demonstrated that these CD133+SMA+ cells can be grown easily in primary cultures from the myocardium of AngII-exposed animals when compared to untreated animals where CD133+SMA+ could not be identified by 3 days in culture. Furthermore, exposure of primary cultures from untreated animals to AngII did not yield expansion of CD133+SMA+ cells supporting our GFP chimera data that the origin of these cells is likely from the BM as opposed to locally proliferating cells.
Fibrocytes have been implicated in liver, kidney, lung and skin fibrosis.20, 39, 40 Furthermore, others have shown that AngII exposure leads to the recruitment of fibrocytes into renal tissue in a model of kidney fibrosis.40 Importantly, fibrocytes can be recruited in response to SDF-1α gradients.20 However, until recently few investigators have suggested a major role for fibrocytes in cardiac fibrosis. Haudek et al41 in a recent publication was able to show that fibroblast precursors can be seen in animals exposed to AngII who develop myocardial fibrosis. The identified fibroblast precursors exhibited leukocyte markers (CD45), fibroblast markers (SMA) and progenitor cell markers (CD34) which, are all in keeping with the phenotype of fibrocytes we have shown in the present manuscript (CD133+/SMA+).41 Our findings complement this recent work supporting the role of fibrocytes and confirming in vivo (GFP chimera) and in vitro (CD133+SMA+ primary cultures) that these cells appear to be fibroblast progenitor cells originating from the BM.
Taken together, our findings support the hypothesis that fibrocytes are the hematopoietic progenitor cell type that is recruited to the myocardium early after AngII exposure. We are proposing that fibrocytes, as opposed to solely inflammatory cells, are a key effector cell in the initiation of myocardial fibrosis. The regulation of fibrocyte recruitment is just beginning to be characterized and is potentially different from that of leukocytes. Thus, additional investigation into the role and regulation of fibrocytes is required to further characterize their role in myocardial fibrosis.
References
Kim S, Iwao H . Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 2000;52:11–34.
Wynn TA . Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214:199–210.
Brasier AR, Recinos III A, Eledrisi MS . Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol 2002;22:1257–1266.
Zhao Q, Ishibashi M, Hiasa K, et al. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension 2004;44:264–270.
Billet S, Aguilar F, Baudry C, et al. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int 2008;74:1379–1384.
Schmieder RE, Hilgers KF, Schlaich MP, et al. Renin-angiotensin system and cardiovascular risk. Lancet 2007;369:1208–1219.
Harada K, Komuro I, Shiojima I, et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998;97:1952–1959.
Li H-L, She Z-G, Li T-B, et al. Overexpression of myofibrillogenesis regulator-1 aggravates cardiac hypertrophy induced by angiotensin II in mice. Hypertension 2007;49:1399–1408.
Liu J, Yang F, Yang X-P, et al. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol 2003;23:776–782.
Mann DLMD . Mechanisms and models in heart failure: a combinatorial approach. Circulation 1999;100:999–1008.
Kawano S, Kubota T, Monden Y, et al. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol 2006;291:H1337–H1344.
Kiarash A, Pagano PJ, Tayeh M, et al. Upregulated expression of rat heart intercellular adhesion molecule-1 in angiotensin II- but not phenylephrine-induced hypertension. Hypertension 2001;37:58–65.
Liu F, Levin MD, Petrenko NB, et al. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J Mol Cell Cardiol 2008;45:715–723.
Crabos M, Roth M, Hahn AW, et al. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J Clin Invest 1994;93:2372–2378.
Dostal DE, Booz GW, Baker KM . Angiotensin II signalling pathways in cardiac fibroblasts: conventional versus novel mechanisms in mediating cardiac growth and function. Mol Cell Biochem 1996;157:15–21.
Sadoshima J, Izumo S . Molecular characterization of angiotensin II—induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res 1993;73:413–423.
Zhou G, Kandala JC, Tyagi SC, et al. Effects of angiotensin II and aldosterone on collagen gene expression and protein turnover in cardiac fibroblasts. Mol Cell Biochem 1996;154:171–178.
Abe R, Donnelly SC, Peng T, et al. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol 2001;166:7556–7562.
Phan SH . Fibroblast phenotypes in pulmonary fibrosis. Am J Respir Cell Mol Biol 2003;29 (3 Suppl):S87–S92.
Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 2004;114:438–446.
Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 2007;13:952–961.
Endo J, Sano M, Fujita J, et al. Bone marrow derived cells are involved in the pathogenesis of cardiac hypertrophy in response to pressure overload. Circulation 2007;116:1176–1184.
Haudek SB, Trial J, Xia Y, et al. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci USA 2008;105:10179–10184.
Underwood RA, Gibran NS, Muffley LA, et al. Color subtractive-computer-assisted image analysis for quantification of cutaneous nerves in a diabetic mouse model. J Histochem Cytochem 2001;49:1285–1291.
Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol 2004;172:4410–4417.
Tacke F, Randolph GJ . Migratory fate and differentiation of blood monocyte subsets. Immunobiology 2006;211:609–618.
Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol 2006;168:659–669.
Moore BB, Kolodsick JE, Thannickal VJ, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol 2005;166:675–684.
Haudek SB, Cheng J, Du J, et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 2010;49:499–507.
Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG . The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 2010;48:504–511.
Varda-Bloom N, Leor J, Ohad DG, et al. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J Mol Cell Cardiol 2000;32:2141–2149.
Zamilpa R, Lindsey ML . Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol 2010;48:558–563.
Alexis D, Christine C, Giulio G . Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 2005;13:7–12.
Reininger AJ . VWF attributes -- impact on thrombus formation. Thromb Res 2008;122 (Supplement 4):S9–S13.
Handgretinger R, Gordon PR, Leimig T, et al. Biology and plasticity of CD133+ hematopoietic stem cells. Annals of the New York Academy of Sciences (HEMATOPOIETIC STEM CELLS 2002: GENETICS AND FUNCTION: Fourth International Symposium) 2003;996:141–151.
Mohle R, Bautz F, Rafii S, et al. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 1998;91:4523–4530.
Bellini A, Mattoli S . The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest 2007;87:858–870.
Bucala R . Circulating fibrocytes: cellular basis for NSF. J Am Coll Radiol 2008;5:36–39.
Moreno M, Ramalho LN, Sancho-Bru P, et al. Atorvastatin attenuates angiotensin II-induced inflammatory actions in the liver. Am J Physiol Gastrointest Liver Physiol 2009;296:G147–G156.
Sakai N, Wada T, Matsushima K, et al. The renin-angiotensin system contributes to renal fibrosis through regulation of fibrocytes. J Hypertens 2008;26:780–790.
Haudek SB, Cheng J, Du J et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 2010;49:499–507.
Acknowledgements
We thank Tanya Myers and Mary Li for their skillful technical assistance. The present work was supported in part by a grant from the Nova Scotia Health Research Foundation (#42311).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sopel, M., Rosin, N., Lee, T. et al. Myocardial fibrosis in response to Angiotensin II is preceded by the recruitment of mesenchymal progenitor cells. Lab Invest 91, 565–578 (2011). https://doi.org/10.1038/labinvest.2010.190
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.190
Keywords
This article is cited by
-
Fabry Disease: Cardiac Implications and Molecular Mechanisms
Current Heart Failure Reports (2024)
-
Effects of apelin on the cardiovascular system
Heart Failure Reviews (2015)
-
Statins Impair Survival of Primary Human Mesenchymal Progenitor Cells via Mevalonate Depletion, NF-κB Signaling, and Bnip3
Journal of Cardiovascular Translational Research (2015)
-
Antibody therapy can enhance AngiotensinII-induced myocardial fibrosis
Fibrogenesis & Tissue Repair (2014)
-
Human mesenchymal stem cells express a myofibroblastic phenotype in vitro: comparison to human cardiac myofibroblasts
Molecular and Cellular Biochemistry (2014)
