
Abstract
Cumulative data obtained from two relatively large pedigrees of a unique reciprocal chromosomal translocation (RCT) t(1;11)(p36.22;q12.2) ascertained by three miscarriages (pedigree 1) and the birth of newborn with hydrocephalus and myelomeningocele (pedigree 2) were used to estimate recurrence risks for different pregnancy outcomes. Submicroscopic molecular characterization by fluorescent in situ hybridization (FISH) of RCT break points in representative carriers showed similar rearrangements in both families. Meiotic segregation patterns after sperm analysis by three-color FISH of one male carrier showed all possible outcomes resulting from 2:2 and 3:1 segregations. On the basis of empirical survival data, we suggest that only one form of chromosome imbalance resulting in monosomy 1p36.22→pter with trisomy 11q12.2→qter may be observed in progeny at birth. Segregation analysis of these pedigrees was performed by the indirect method of Stengel-Rutkowski and showed that probability rate for malformed child at birth due to an unbalanced karyotype was 3/48 (6.2±3.5%) after ascertainment correction. The risk for stillbirths/early neonatal deaths was −/48 (<1.1%) and for miscarriages was 17/48 (35.4±6.9%). However, the probability rate for children with a normal phenotype at birth was 28/48 (58.3±7.1%). The results obtained from this study may be used to determine the risks for the various pregnancy outcomes for carriers of t(1;11)(p36.22;q12.2) and can be used for genetic counseling of carriers of this rearrangement.
Similar content being viewed by others
Introduction
Carriers of reciprocal chromosomal translocations (RCTs) may be at risk for abnormal pregnancy outcomes including offspring with developmental disabilities and malformations, recurrent miscarriages and/or stillbirths/early neonatal death. Depending on the rearrangement, RCT carriers are at risk of producing different gametes with unbalanced karyotypes, each of which will have different survival rates.1,2 In the absence of large pedigrees, to understand the pattern of the meiotic segregation of individual RCT carriers, the study of sperm cells is necessary.1, 2, 3, 4 The frequencies of particular forms of unbalanced gametes are dependent on factors including the size of the chromosomal segments, the genetic content, location of break points and survival of certain imbalances, and are characteristic for individual RCT.1,2 The collection of relatively numerous empirical and cytogenetic data of individual RCT carrier pedigrees is necessary to ascertain the risks of malsegregation associated with particular translocations.5, 6, 7, 8, 9
Because the risk of malsegregation is dependent on the size and chromosomal origins of the segments involved and placement of the break points, the accuracy of identifying the break point positions for particular RCT is crucial. High-resolution molecular cytogenetic techniques, like metaphase, interphase and fiber fluorescent in situ hybridization (FISH), sequence-tagged site (STS) marker walking and sequencing of chromosomal regions containing the rearrangement break points and junctions, may be useful for better characterization of specific rearrangements.10, 11, 12, 13, 14
Here, we present results of segregation analysis of two unrelated pedigrees carrying similar rearrangements t(1;11)(p36.22;q12.2) and meiotic segregation patterns after sperm analysis of one male carrier.
Materials and methods
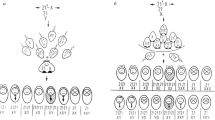
The study group consisted of the collected empirical and cytogenetic data of two pedigrees with carriers of a t(1;11)(p36.22;q12.2). To the best of our knowledge, these families are unrelated. The families were ascertained either because of three miscarriages (Figure 1a—pedigree 1, IV:2–4) or owing to the death of a two-day-old newborn with many malformations, including hydrocephalus and myelomeningocele (Figure 1b—pedigree 2, IV:3).

Pedigrees data: (a) pedigree 1; (b) pedigree 2 (pedigree was drawn according to Bennett et al.49).
Cytogenetic study
Conventional Cytogenetics
Cell cultures from peripheral lymphocytes were performed according to standard procedures. Chromosomes were stained by GTG and RBG banding techniques, with resolution of 550–850 bands/haploid set.
The semen samples from a male carrier from pedigree 2 (Figure 1b—III:13), after ejaculation and washing in PBS, were fixed according to classical cytogenetic methods.
Informed written consents were obtained from all participating individuals after explanation of possible consequences of the study, in accordance with the Declaration of Helsinki. The research protocols were approved by the Institutional Review Boards at both Medical University of Bialystok and Poznan University of Medical Sciences.
Fluorescent in situ hybridization of chromosomes from lymphocytes
FISH was performed with probes corresponding to the break point clones on the 1p36 as previously described.11 In addition, bacterial artificial chromosome (BAC) probes were selected using the UCSC [www.genome.ucsc.edu] and NCBI [www.ncbi.nlm.nih.gov] genome browsers to identify clones to 11q. These clones were used in metaphase FISH to determine the size of the translocated chromosome segments (Figure 2, Supplementary Figure 1).

Break point-positions localization on the chromosomes involved in the balanced chromosomal translocation t(1;11)(p36.22;q12.2) (pedigree 1, III:15): (a) partial karyotype studied by GTG and RBG techniques with indication of break point positions on chromosomes and ideograms; (b) break point position using BAC clones (FISH technique) on 1p36.22 mapped between two clones BAC: RP11-575L21 and RP11-874A11, top—hybridization signal of RP4-575L21 (red) visible on 1 and der(11) chromosomes, signal of RP11-799F14 (green) visible on 11 and der(11) chromosomes; bottom—signal of RP11-498B2 (red) visible on 1 and der(11) chromosomes, signal of RP11-874A11 (green) visible on 1 and der(1) chromosomes; and (c) break point position using FISH on 11q12.2 mapped at RP11-881M11 BAC clone, top—hybridization signal of RP4-575L21 (red) visible on 1 and der(11) chromosomes, signal of RP11-799F14 (green) visible on 11 and der(11) chromosomes, middle—signal of RP11-881M11 (red) visible on 11, der(11) and der(1) chromosomes, signal of RP11-799F14 (green) visible on 11 and der(11) chromosomes, bottom—hybridization signal of RP11-496H15 (red) visible on 1 and der(11) chromosomes, signal of RP11-855O10 (green) visible on 11 and der(1) chromosomes.
FISH mapping
FISH mapping of break point positions was carried out on metaphase chromosomes using the following BAC clones covering the 1p36 region: RP5-1115A15 [AL096855; chr1:8 387 143–8 565 545], RP11-476D13 [AL358252; chr1:9 349 187–9 373 344], RP11-498B2 [AC013728; chr1:9 762 329–9 934 477], RP11-807G9 [AL603962; chr1:9 953 249–9 983 505], RP11-496H15 [AL590639; chr1:9 981 506–10 077 581], RP4-575L21 [AL096841; chr1:10 075 582–10 170 003], RP11-874A11 [AP001098; chr1:9885646–11218505] and the 11q12.2 region: RP11-799F14 [genomic alignments of BAC ends: AQ525174 and AQ556193; chr11:58 039 510–58 040 025 and chr11:58 231 103–58 231 589, respectively], RP11-881M11 [AP003721; chr11:60 417 795–60 621 331], RP11-855O10 [ENST00000506329, ENST00000542121; chr11:61 355 966–61 375 022 and chr11:61 422 137–61 423 174, respectively], in accordance with Human March 2006 (NCBI36/hg18) Assembly.
Generation of somatic cell hybrids
To identify the 1p36 break point, somatic cell hybrids from subject 1p112c were generated as previously described.15 Transformed lymphoblastoid cells from our subject were fused to hypoxanthine guanine phosphoribosyl transferase-deficient RJK88 hamster cells. Somatic cell hybrids containing either the derivative chromosome 1 or the normal chromosome 1 were identified.
STS marker walking, amplification across the junction and junction DNA sequencing
To identify the 1p36 and 11q12.2 break points in subject 1p112c, (Figure 1a–III:15), STS markers were designed and polymerase chain reaction analyses were performed using hybrid DNA containing the derivative chromosome 1. DNA sequences obtained from GenBank [www.ncbi.nlm.nih.gov] corresponding to the genomic regions containing the 1p36 and 11q12.2 break points were analyzed. Repetitive elements were masked from the sequence using Repeat Masker [www.repeatmasker.org] and 200–400 bp STS markers were designed from the unmasked unique sequence using Primer 3 [http://Frodo.wi.mit.edu/cgi-bin]. Break point at 1p36 on derivative chromosome 1 was narrowed using markers spaced ~20 kb apart along the length of the chromosome followed by ~5 kb apart.
Using several pairs of primers at 11q12.2, the breakpoint was narrowed to 2.5 kb according to the method of break point identification described previously by Gajecka et al.14 Then, in order to identify the t(1;11) junction, amplification across the junction was designed with sets of unique-sequence reverse primers located at 11q12.2 and forward primers from the putative 1p36 break point regions. Obtained products of polymerase chain reactions were cut from agarose gel and purified. Then, sequencing polymerase chain reactions before Sanger sequencing were carried out using the same sets of unique-sequence reverse primers located at 11q12.2 and forward primers from the putative 1p36 break point regions. Obtained polymerase chain reaction products containing the putative junctions were sequenced using an ABI PRISM 3100-Avant genetic analyzer and Big Dye reagents (Applied Biosystems, Inc., Foster City, CA, USA). Sequencher 4.6 (Gene Codes) was used to analyze sequences.
FISH procedure to sperm analysis
A three-color FISH method according to the manufacturer’s protocol was performed. Directly labeled probes (Aquarius, Cytocell Technologies Ltd., Cambridge, UK) were used; combination of red and green α-satellite probes of chromosome 11 (locus D11Z1), namely 11c Red and 11c Green, to give a yellow color, and red and green subtelomere probes, namely 1p Red and 11q Green. To determine X:Y ratio, α-satellite probes of chromosomes X (Xc Green, locus DXZ1) and Y (Yq12 Red, locus DYZ1) were used.
The assessment of meiotic segregation patterns
To analyze the meiotic segregation patterns, the expected quadrivalent configuration of the translocation at meiosis I was constructed with the positions of FISH probes shown (see Supplementary Figure 2).
The assessment of segregation pattern for different categories of pregnancy outcome
On the basis of segregation analysis, the probability rates of different categories of pregnancy outcomes (that is, birth of malformed child probably due to unbalanced karyotype, stillbirth/early neonatal death and miscarriages) and child without malformations have been calculated according to the method of Stengel-Rutkowski et al.16 and Stene and Stengel-Rutkowski.17 Ascertainment correction, elimination of probands and their parents, was calculated according to the method of Stene and Stengel-Rutkowski17 and recently proposed by Gardner et al.18 The probability estimates for unfavorable pregnancies were calculated according to the following:

p—risk value of different pregnancy outcome (probability rate of malformed child at birth, or risk of stillbirth/ neonatal death or risk of miscarriages, a—number of unfavorable pregnancies after ascertainment correction, n—number of all pregnancies after ascertainment correction, S—s.d.
pmax—maximum risk, e—2.71828 is the base number for natural logarithms
The empirical data were sufficiently large to allow for direct segregation analysis of pedigrees, enabling the calculation of the probability rates for unbalanced offspring at birth, for stillbirths/neonatal deaths and miscarriages.
Results
Cytogenetic study
Karyotype analysis of a proband from pedigree 1 (III:15) revealed the RCT t(1;11)(p36;q12) and similar RCT carriers involving chromosomes 1p and 11q at the same cytological break point positions in the father of the proband (III:4) of pedigree 2. The interpretation of break point positions is shown in Figure 2a.
Using FISH, a carrier (III:15) from pedigree 1 revealed the subterminal localization of the break point position at chromosome 1, mapped between two BAC clones: RP4-575L21 and RP11-874A ~10.2–10.5 Mb from 1pter (Figure 2b). The break point position for chromosome 11 was mapped using BAC clone RP11-881M11 ~60.5 Mb from 11qter (Figure 2c). The karyotype was then reinterpreted as: t(1;11)(p36.22;q12.2). ish t(1;11)(RP5-1115A15−,RP11-476D13−,RP11-498B2−,RP11-807G9−,RP11-496H15−,RP4-575L21−,RP11-874A11+;RP11-855O10−,RP11-881M11+,RP11-799F14+). Thus, based on this analysis, carriers of this particular translocation are predicted to be at risk for double-segment imbalances in offspring. Based on FISH results completed for a carrier from pedigree 1, (Figure 1a—III:15) similar FISH experiments were performed for carriers from pedigree 2 (Figure 1b—II:2, III:13, Supplementary Figure 1). The same hybridization pattern was found in all carriers, indicating similar rearrangement. STS marker walking analyses using hybrid containing derivative chromosomes 1 did not help to define the 1p36 and 11q12.2 break points in these rearrangements at the sequence level due to repetitive elements at the break points. The break point region at derivative 1 was narrowed by STS marker walking to a sequence fragment consisting of >3 kb of repetitive elements and the break point at 11q12.2 contained at least 2.5 kb of repetitive elements. Chromosomal position of 1p36 break point was narrowed to chr1:10 172 755–10 176 276 region (3522 bp), whereas the 11q break point to chr11:60 551 802–60 559 898 (8097 bp) chromosomal area (hg18) using primers listed in Supplementary Table 1. The presence of repetitive elements at the break points prohibited further break point narrowing and amplification across the junctions, which was unsuccessful although several sets of primers were used.
Family studies
Pedigree 1
A familial RCT was ascertained in 27-year-old woman (III:15) from a couple studied because of three miscarriages (IV:2–4) (Figure 1a). Further family studies revealed additional carriers of the same balanced RCT in her father (II:12), two uncles (II:4, II:10) and in one of the uncle’s son (III:9). Four pregnancies resulted in miscarriage (III:10–13) in proband’s parents and one miscarriage (III:2) in her uncle (II:4).
Pedigree 2
This familial RCT was ascertained in 34-year-old father (III:4) investigated because of early death of a 2-day-old girl (IV:3) with malformations including hydrocephalus with myelomeningocele (Figure 1b). Further family studies revealed that this RCT occurred in her aunt (III:11) and two children (IV: 10, IV:15), her uncle (III:13), son of her uncle (IV:17), grandmother (II:2) and of grandmother’s sister (II:10). The developmental malformations of the nervous system have been observed also in the sister (III:8) of the father. Unfortunately she died at 20 weeks of age and she was not karyotyped. In addition, a similar phenotype with myelomeningocele and hydrocephalus included has been observed in the other female relative from the grandmother’s side (II:7, II:12, III:30). Several miscarriages were observed in each family from both pedigrees, including the parents of proband (IV:1,5–6), in family of an aunt (IV:11–14), family of an uncle (IV:18), grandparents of proband (III:1–3) and grandmother’s sister (II;10).
Segregation analysis of pedigrees
Ascertainment correction
Among 23 pregnancies in pedigree 1, five pregnancies have been omitted after ascertainment correction: three miscarriages (IV:2–4), proband (III:15) and her father (II:12). Among 33 pregnancies in pedigree 2, three pregnancies were omitted after ascertainment correction: proband (IV:3), her father (III:4) and grandmother (II:2). Finally, there were 48 pregnancies as a total number after ascertainment correction used in the cumulative data.
Malformed child at birth probably due to unbalanced karyotype
As karyotype data were not available from the children with developmental malformations born at term and with limited survival rate, we used the pedigree information to calculate the rate for malformed child at birth probably due to karyotype imbalance. For pedigree 1, this probability rate was −/18 and for pedigree 2 was 3/30 after ascertainment corrections. Cumulative data from the two pedigrees showed that the probability rate of having an unbalanced child with malformations at birth for carriers of this translocation was 3/48 (6.2±3.5%) (Table 1).
Stillbirths/neonatal deaths
In families from pedigree 1 and pedigree 2, no stillbirth/early neonatal death among 48 pregnancies after ascertainment correction was found. Using data from both pedigrees, we demonstrated that the risk for stillbirths/early neonatal deaths after ascertainment correction was −/48 (<1.1%) (Table 1).
Miscarriages
Eight miscarriages were observed in pedigree 1. Three of them were omitted for ascertainment correction (Figure 1a—IV:2–4), resulting in a total of 18 pregnancies after ascertainment correction. The risk for miscarriages after ascertainment correction was 5/18 (28±13%). For pedigree 2, 12 miscarriages among 30 pregnancies were used for the calculations after ascertainment correction. Hence, the rate for miscarriages in pedigree 2 was 12/30 (40±8.9%). Using data from two pedigrees, 48 pregnancies and 17 miscarriages after ascertainment correction were used to calculate the risk resulting in 17/48 (35.4±6.9%) (Table 1).
Parental origin of t(1;11)(p36.22;q12.2)
We found three maternal carriers (Figure 1a—III:15; Figure 1b—III:11, II:2; II;10) and five paternal carriers (Figure 1a—II:4,10,12; Figure 1b—III:4,13) of this translocation. In two pedigrees in which the karyotypes were unknown (Figure 1a–I:1,2; Figure 1b–I: 1,2,3), the carrier state could be established on the basis of pedigree analysis. For female carriers, the probability rate for a malformed child at birth was 1/16 (6.2±6.1%), for stillbirths/early neonatal death was −/16 (<2.9%), and for miscarriages was 8/16 (50±12.5%) after ascertainment correction. For male carriers, the probability rate for malformed child at birth was −/19 (<2.6%), for stillbirths/early neonatal deaths was also −/19 (<2.6%) and for miscarriages was 9/19 (47.4±11.5%) after ascertainment correction (Table 2).
Child without malformations
In pedigree 1, 13 children without malformations among a total number of 18 pregnancies after ascertainment correction were identified. Therefore, the probability rate for this pregnancy outcome was 13/18 (72.0±8.5%) (Figure 1a, Table 3). In pedigree 2, 15 children without malformations among 30 pregnancies after ascertainment correction were identified. Thus, the probability rate for this pregnancy outcome was 15/30 (50±9.1%) (Figure 1b, Table 3). Using data from two pedigrees, 48 pregnancies and 28 children without malformations after ascertainment correction were identified. Finally, the probability rate for children without malformations at birth was 28/48 (58.3±7.1%) (Table 3).
Meiotic segregation pattern
The frequencies of different types of sperm segregation are listed in Supplementary Table 1. In total, 3151 sperm cell nuclei were used for meiotic segregation analysis, and 5971 for X:Y ratio were analyzed with efficiency of FISH hybridization of 98%. The frequencies of alternate, adjacent I, adjacent II and 3:1 segregations were 40.4, 37.5, 5.4 and 11.3%, respectively. All possible outcomes after 2:2 and 3:1 segregations were observed (Supplementary Figure 2, Supplementary Table 2). The most common segregation types were alternate and adjacent I (similar frequencies). A low number was observed for adjacent II, which was in contrast to 3:1 segregation, which showed a high proportion of unbalanced gametes. The color patterns for normal and balanced karyotypes were indistinguishable, so we are not able to determine the ratios for these possibilities. Atypical signals in 5.4% of spermatozoa were found. Probable recombination signals can be inferred from two yellow signals and no green or red, whereas lack of any signals could be the result of a hybridization failure. The frequencies of spermatozoa with chromosome X or Y was 51.8 and 47.5%, respectively. The incidences of spermatozoa bearing two chromosomes XX, YY or XY were: 0.0837%, 0.0502% and 0.5024%, respectively, of which the first two are within the normal range of normozoospermic samples (n=7 controls), whereas the XY hyperhaploidy was significantly higher in the carrier as compared with the control value of 0.08 (P<0.05).
Discussion
Unique RCT and the risk for double segment imbalance in progeny
We described carriers of an unique familial chromosomal reciprocal translocation t(1;11)(p36.22;q12.2) in nine Polish families from two pedigrees. Although these families are living in the same region (Podlasie), we could not establish that they are related. These families presented with multiple miscarriages or with a newborn with malformations, including hydrocephalus and myelomeningocele. Detection of the break point position of the RCT in 1p was subterminal, at 1p36.22 determined by FISH using BAC clones. The location of this break point allowed us to presume that these carriers of t(1;11)(p36.22;q12.2) would be at risk for double segment imbalances in the progeny. The 1p36 region is characterized by molecular instability resulting in terminal and interstitial deletions, complex rearrangements and unbalanced translocations.11,14 The use of new genomic methods opens up the possibilities to find more examples of RCT involving this region of 1p36. In this study, the application of FISH using BAC probes was helpful for determining the break points and resulted in a revision of both break point positions (terminal or subterminal in 1p and in defining the 11q breakpoint).
In our search of the literature, we did not find any other publications describing this particular RCT between segment 1p36.22p36.3 and 11q12.2→qter, although there are several other case reports of translocations involving other segments of 11q or other chromosomes translocated with 1p36-pter.11,19, 20, 21, 22, 23 Similarly, a shorter segment involving 11q13→qter band found in a RCT has been observed.24, 25, 26
Meiotic segregation pattern
To identify the various possible genetic imbalances that can be produced by carriers of chromosomal translocations, the meiotic segregation patterns in sperm cells can be evaluated. To date, the meiotic segregation patterns in sperm cells from >200 different RCTs have been studied.3,4,27,28 One case of an RCT carrier with similar (but not identical) break points [t(1;11)(p36.3;q13.1)] to our translocation [t(1;11)(p36.22;q12.2)] was analyzed and described in three different studies.29, 30, 31 Spriggs et al.30 used the in vitro human sperm penetration assay with total of 548 sperm karyotypes identified. Spriggs and Martin31 analyzed segregation pattern on sperm cells by using two-color FISH with centromeric probes (n=13 061). In the study of Goldman and Hulten,29 FISH was used with paracentromeric probes to evaluate the expected gametic proportions from the meiotic analysis (including number of chiasmata and quadrivalent behavior) in germ cells (n=80) obtained from testicular biopsy. Results obtained in this study were similar to the previously published report of Spriggs et al.30 in which sperm meiotic segregation patterns were analyzed after spermatozoa penetrated the oocyte thus disclosing their karyotypes. The results reported by Goldman and Hulten29 and Spriggs et al.30 were as follows: alternate 38.1/33.2%, adjacent I 39.4/42.9%, adjacent II 13.75/15.9% and 3:18.75/8.00%, respectively. When comparing results of Goldman and Hulten29 with the third study of Spriggs and Martin,31 marked differences in proportion of adjacent II segregants were observed (13.75% vs 8.3%, respectively). Moreover, our results for adjacent II segregation type (5.4%) (Supplementary Table 1) were similar to this study.30 To compare our results with the observations obtained by Spriggs and Martin,31 we summed the frequencies of alternate and adjacent I segregation outcomes. Because Spriggs and Martin31 used a two-color FISH, they could not identify which sperm were the result of alternate or adjacent I segregation—normal chromosome and its derivative contained the same centromeric sequences (82.5% vs 77.8%—in our study). Also in the case of 3:1 segregation, there were no differences in gamete frequencies between our results and that obtained by Spriggs and Martin31 (9.2% vs 11.3% obtained in our study). On the basis of our results of meiotic segregation patterns, we can see that all forms of meiotic malsegregation are produced by our RCT carrier of t(1;11)(p36.22;q12.2).
Comparison of meiotic segregation patterns and segregation analysis of empiric data from two pedigrees
We speculated that progeny with almost all possible outcomes of karyotypic imbalances probably have limited survival. Indeed, among 48 pregnancies of carriers, there have been 17 recognizable miscarriages, four malformed children at birth probably due to unbalanced karyotype. After ascertainment correction, the probability rate of miscarriages for carriers of t(1;11)(p36.22;q12.2) was high, 17/48 (35.4±6.9%). The high number of miscarriages detected in comparison with the high rate of unbalanced gametes (54%) suggests a strong selection against the unbalanced karyotypes during fetal development. This assumes that the ability for fertilization has been preserved in any unbalanced spermatozoa (54%).2 Unfortunately, we were not able to karyotype any of the miscarriages, all presumed to be unbalanced for the purpose of our risk calculations. It is possible that the fertilized eggs with unbalanced translocation were negatively selected before noticing pregnancy, which is sometimes manifested as a delayed menstruation. This type of information was not available in the examined families. However, in both studied families, in II:1 in pedigree 1 (Figure 1a) and II:5 in pedigree 2 (Figure 1b), the offspring was not observed. Because the material was not available for karyotype testing, the diagnosis of infertility of unknown reasons was concluded.
Hydrocephalus, myelomeningocele and other malformations of the nervous system in the phenotype of five unkaryotyped children have been observed. On the basis these clinical observations, we can speculate that they resulted from chromosome imbalances because of the similar phenotypes observed in children at birth monosomy 1p36 or trisomy 11q13→qter.21,24, 25, 26,32, 33, 34, 35, 36, 37 Therefore, we can presume that five children (Figure 1b—IV:3, III:8, II:7 and II:12, III: 30) with hydrocephalus and myelomeningocele observed in pedigree 2 could represent the phenotypic effects of the unbalanced karyotype resulting in monosomy 1p36.22→pter with trisomy 11q12.2→qter. However, because of the lack of karyotypic evidence, additional cases must be ascertained and karyotyped to prove our hypothesis. Monosomy 11q12.2→qter or trisomy 11q12.2→cen→pter have not been observed so far and is likely not compatible with survival until birth, therefore taking into consideration the phenotypic effects of the chromosome imbalance, we suggest that only monosomy 1p36.22→pter with trisomy 11q12.2→qter, may be observed in malformed progeny at birth with presumably limited survival after delivery.
Size of imbalanced segments and survival rate
1p36-pter region
Monosomy 1p36 syndrome is one of the most commonly occurring contiguous gene syndrome. The size of the imbalance is variable with 1p36 deletions arising de novo or resulting from malsegregation of a parental RCT.38, 39, 40, 41 The clinical findings in our pedigrees of neonatal have been reported with monosomy 1p3619,32,34,35,38,40, 41, 42 as well as reported with trisomy 11q13.26 Monosomy of 1p36 of ~16 Mb in size was described by Nicoulaz et al.35 in a child born at 31 week of gestation who passed away at 48 h after delivery. It is interesting that, in general, 10–11 Mb deletions of 1p36 typically do not result in lethality.39,42, 43, 44 However, deletions over 11–16 Mb may be lethal.36,37 As in our case, unbalanced segregants that result in double segment imbalances will impact the survival rate and is dependent on the size of the two segments and genetic content. There is usually a higher rate for complex malformations, which may result in early lethality, in comparison with single segment imbalances.7,16
Our observation confirms two previous suggestions of Saito et al.37 and Nicoulaz et al.35 that 1p36 region for pure distal terminal deletions, >11 Mb is the haplolethal. Although many large 1p36 deletions ~10–11 Mb in size are likely nonlethal,39,42, 43, 44 additional observations are necessary to determine if monosomy 1p36-pter sized >11 Mb is risk factor for limited survival rate.
11q12.2-qter region
According to our knowledge, there are no reports of offspring with pure trisomy 11q12.2.→qter to determine if such type of imbalance would be a risk factor for limited survival at birth. It should be emphasized that the resolution of banding techniques used for evaluation of break point positions is limited; in many cases, break points in either 11q13 and 11q12.2 bands cannot be distinguished. Liveborn children with trisomy 11q13→qter have been observed.24, 25, 26 However, Brewer et al.45 after analysis of data from Human Cytogenetics Database, proposed that band 11q12 is haplo- and triplolethal. Thus, the presence of 11q12.2→qter in double segment imbalances likely contributes to limited survival at birth. Among others, there are genes that may be contributing to the phenotypes observed in our cases. The gene HGNC is responsible for hydrolethalus syndrome and was assigned to 11q24.2 band46,47 and FOLR1, a gene located at 11q13.4 band whose overexpression was observed in neural tube defects.48
Parental origin of RCT carrier and value of probability estimation
There were no differences (Table 2) in risk values of different categories of pregnancy outcomes dependent on paternal or maternal origin of RCT carriership. The such type of differences in risk value may be expected especially in case the prevalence of production of gametes after 3:1 disjunction and interchange segregation.16 Our results of meiotic segregation pattern show high frequencies of alternate (40.4%) and adjacent I (37.5%) segregations in comparison with low frequency of 3:1 (11.3%) segregations. It explains any differences between risk values obtained from pedigree data.
Risk estimates of different pregnancy outcomes for carriers of t(1;11)(p36.22;q12.2) and the results of meiotic segregation analysis may be used for genetic counseling of such RCT carriers. The analysis of meiotic segregation patterns in sperm provides information about the types of unbalanced gametes, which may be generated in male carriers of RCT. Survival rate of progeny with different forms of unbalanced karyotypes in the perinatal period varies depending on the particular form of imbalance. Therefore, the results of meiotic segregation pattern together with the empiric data from pedigrees supplement each other.
References
Midro AT, Wiland E, Panasiuk B, Leśniewicz R, Kurpisz M Risk evaluation of carriers with chromosome reciprocal translocation t(7;13)(q34;q13) and concomitant meiotic segregation analyzed by FISH on ejaculated spermatozoa. Am. J. Med. Genet. Part A 140, 245–256 (2006).
Wiland E, Midro AT, Panasiuk B, Kurpisz M The analysis of meiotic segregation patterns and aneuploidy in the spermatozoa of father and son with translocation t(4;5)(p15.1;p12) and the prediction of the individual probability rate for unbalanced progeny at birth. J. Androl. 28, 262–272 (2007).
Benet J, Oliver-Bonet M, Cifuentes P, Templado C, Navarro J Segregation of chromosomes in sperm of reciprocal translocation carriers: a review. Cytogenet. Genome Res. 111, 281–290 (2005).
Perrin A, Morel F, Douet-Guilbert N, Le Bris MJ, Amice J, Amice V et al. A study of meiotic segregation of chromosomes in spermatozoa of translocation carriers using fluorescent in situ hybridisation. Andrologia 42, 27–34 (2010).
Engels H, Eggermann T, Caliebe A, Jelska A, Schubert R, Schüler HM et al. Genetic counseling in Robertsonian translocations der(13;14): frequencies of reproductive outcomes and infertility in 101 pedigrees. Am. J. Med. Genet. Part A 146, 2611–2616 (2008).
Midro AT, Stengel-Rutkowski S, Stene J Experiences with risk estimates for carriers of chromosomal reciprocal translocations. Clin. Genet. 41, 113–122 (1992).
Midro AT, Panasiuk B, Stasiewicz-Jarocka B, Iwanowski PS, Fauth C, Speicher MR, Lesniewicz R Risk estimates for carriers of chromosome reciprocal translocation t(4;9)(p15.2;p13). Clin. Genet. 58, 153–155 (2000).
Panasiuk B, Usinskiené R, Kostyk E, Rybałko A, Stasiewicz-Jarocka B, Krzykwa B et al. Genetic counselling in carriers of reciprocal chromosomal translocations involving short arm of chromosome X. Ann. Genet. 47, 11–28 (2004).
Stasiewicz-Jarocka B, Haus O, Van Assche E, Kostyk E, Constantinou M, Rybałko A et al. Genetic counseling in carriers of reciprocal chromosomal translocations involving long arm of chromosome 16. Clin. Genet. 66, 189–207 (2004).
Gajecka M, Glotzbach CD, Jarmuz M, Ballif BC, Shaffer LG Identification of cryptic imbalance in phenotypically normal and abnormal translocation carriers. Eur. J. Hum. Genet. 14, 1255–1262 (2006).
Gajecka M, Glotzbach CD, Shaffer LG Characterization of a complex rearrangement with interstitial deletions and inversion on human chromosome 1. Chromosome Res. 14, 277–282 (2006).
D'Angelo CS, Gajecka M, Kim CA, Gentles AJ, Glotzbach CD, Shaffer LG et al. Further delineation of nonhomologous-based recombination and evidence for subtelomeric segmental duplications in 1p36 rearrangements. Hum. Genet. 125 (5-6), 551–563 (2009).
Gajecka M, Saitta SC, Gentles AJ, Campbell L, Ciprero K, Geiger E et al. Recurrent interstitial 1p36 deletions: Evidence for germline mosaicism and complex rearrangement breakpoints. Am. J. Med. Genet. Part A 152A, 3074–3083 (2010).
Gajecka M, Gentles AJ, Tsai A, Chitayat D, Mackay KL, Glotzbach CD et al. Unexpected complexity at breakpoint junctions in phenotypically normal individuals and mechanisms involved in generating balanced translocations t(1;22)(p36;q13). Genome Res. 18, 1733–1742 (2008).
Page SL, Shaffer LG Nonhomologous Robertsonian translocations form predominantly during female meiosis. Nat. Genet. 15, 231–232 (1997).
Stengel-Rutkowski S, Stene S, Gallano P . Risk Estimates in Balanced Parental Reciprocal Translocations: 147 (Expansion Scientifique Francaise, Paris, 1988).
Stene J, Stengel-Rutkowski S in Cytogenetics of Mammalian Autosomal Rearrangements (ed Daniel, A.) 1–54 (Alan R. Liss Inc: New York, NY, USA, 1988).
Gardner RJM, Sutherland GR, Shaffer LG in Chromosome Abnormalities and Genetic Counseling 4th edn. (eds Gardner, R. J. M., Sutherland G. R., Shaffer L. G.). Ch 4, 59–64 (Oxford University Press, Inc., New York, 2012).
Campeau PM, Ah Mew N, Cartier L, Mackay KL, Shaffer LG, Der Kaloustian VM et al. Prenatal diagnosis of monosomy 1p36: a focus on brain abnormalities and a review of the literature. Am. J. Med. Genet. Part A 146, 3062–3069 (2008).
Chen E, Obolensky E, Rauen KA, Shaffer LG, Li X Cytogenetic and array CGH characterization of de novo 1p36 duplications and deletion in a patient with congenital cataracts, hearing loss, choanal atresia, and mental retardation. Am. J. Med. Genet. Part A 146, 2785–2790 (2008).
Hiraki Y, Fujita H, Yamamori S, Ohashi H, Eguchi M, Harada N et al. Mild craniosynostosis with 1p36.3 trisomy and 1p36.3 deletion syndrome caused by familial translocation t(Y;1). Am. J. Med. Genet. Part A 140, 1773–1777 (2006).
Lennon PA, Cooper ML, Curtis MA, Lim C, Ou Z, Patel A et al. Array-based comparative genomic hybridization facilitates identification of breakpoints of a novel der(1)t(1;18)(p36.3;q23)dn in a child presenting with mental retardation. Am. J. Med. Genet. Part A 140, 1156–1163 (2006).
Midro AT, Stasiewicz-Jarocka B, Leśniewicz R Polish collection of balanced translocations, (Nr 5 Report of Committee of Human Genetics Polish Academy of Sciences, Białystok, 1999).
Kotzot D, Röthlisberger B, Riegel M, Schinzel A Maternal uniparental isodisomy 11q13qter in a dysmorphic and mentally retarded female with partial trisomy mosaicism 11q13qter. J. Med. Genet. 38, 876–881 (2001).
Pihko H, Therman E, Uchida IA Partial 11q trisomy syndrome. Hum. Genet. 58, 129–134 (1981).
Tinsa F, Chebbi Y, Meddeb M, Bousnina D, Boussetta K, Bousnina S Monosomy 10q26-qter and trisomy 11q13-qter as a result of de novo unbalanced translocation. J. Appl. Genet. 50, 289–291 (2009).
Anton E, Vidal F, Blanco J Role of sperm FISH studies in the genetic reproductive advice of structural reorganization carriers. Hum. Reprod. 22, 2088–2092 (2007).
Perrin A, Caer E, Oliver-Bonet M, Navarro J, Benet J, Amice V et al. DNA fragmentation and meiotic segregation in sperm of carriers of a chromosomal structural abnormality. Fertil. Steril. 92, 583–589 (2009).
Goldman ASH, Hulten MA Analysis of chiasma frequency and first meiotic segregation in a human male reciprocal translocation heterozygote, t(1;11)(p36.3;q13.1), using fluorescence in situ hybridization. Cytogenet. Cell Genet. 63, 16–23 (1993).
Spriggs EL, Martin RH, Hulten M Sperm chromosome complements from two human reciprocal translocation heterozygotes. Hum. Genet. 88, 447–452 (1992).
Spriggs EL, Martin RH Analysis of segregation in a human male reciprocal translocation carrier, t(1;11)(p36.3;q13.1), by two-colour fluorescence in situ hybridization. Mol. Reprod. Dev. 38, 247–250 (1994).
Barbi G, Kennerknecht I, Klett C Reciprocal translocation t(1;15)(p36.2;p11.2): confirmation of a suggestive cytogenetic diagnosis by in situ hybridization and clinical case report on resulting monosomy (1p). Am. J. Med. Genet. 43, 722–725 (1992).
Heilstedt HA, Shapira SK, Gregg AR, Shaffer LG Molecular and clinical characterization of a patient with duplication of 1p36.3 and metopic synostosis. Clin. Genet. 56, 123–128 (1999).
Keppler-Noreuil KM, Carroll AJ, Finley WH, Rutledge SL Chromosome 1p terminal deletion: report of new findings and confirmation of two characteristic phenotypes. J. Med. Genet. 32, 619–622 (1995).
Nicoulaz A, Rubi F, Lieder L, Wolf R, Goeggel-Simonetti B, Steinlin M et al. Contiguous ~16Mb 1p36 deletion: Dominant features of classical distal 1p36 monosomy with haplo-lethality. Am. J. Med. Genet. Part A 155, 1964–1968 (2011).
Ribeiro Mdo C, Gama de Sousa S, Freitas MM, Carrilho I, Fernandes I Bilateral perisylvian polymicrogyria and chromosome 1 anomaly. Pediatr. Neurol. 36, 418–420 (2007).
Saito S, Kawamura R, Kosho T, Shimizu T, Aoyama K, Koike K et al. Bilateral perisylvian polymicrogyria, periventricular nodular heterotopia, and left ventricular noncompaction in a girl with 10.5-11.1 Mb terminal deletion of 1p36. Am. J. Med. Genet. A 146, 2891–2897 (2008).
Battaglia A Del 1p36 syndrome: a newly emerging clinical entity. Brain Dev. 5, 358–361 (2005).
Gajecka M, Mackay KL, Shaffer LG Monosomy 1p36 deletion syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 145, 346–356 (2007).
Heilstedt HA, Ballif BC, Howard LA, Kashork CD, Shaffer LG Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Clin. Genet. 64, 310–316 (2003).
Slavotinek A, Shaffer L, Shapira S Monosomy 1p36. J. Med. Genet. 36, 657–663 (1999).
Heilstedt HA, Ballif BC, Howard LA, Lewis RA, Stal S, Kashork CD et al. Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am. J. Hum. Genet. 72, 1200–1212 (2003).
Dobyns WB, Mirzaa G, Christian SL, Petras K, Roseberry J, Clark GD et al. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2. Am. J. Med. Genet. Part A 146, 1637–1654 (2008).
Shapira SK, McCaskill C, Northrup H, Spikes AS, Elder FF, Sutton VR et al. Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am. J. Hum. Genet. 61, 642–650 (1997).
Brewer C, Holloway S, Zawalnyski P, Schinzel A, Fitz Patrick D A chromosomal duplication map of malformations: regions of suspected haplo- and triplo lethality—and tolerance of segmental aneuploidy in humans. Am. J. Hum. Genet. 64, 1702–1708 (1999).
Mee L, Honkala H, Kopra O, Vesa J, Finnilä S, Visapää I et al. Hydrolethalus syndrome is caused by a missense mutation in a novel gene HYLS1. Hum. Mol. Genet. 14, 1475–1488 (2005).
Visapää I, Salonen R, Varilo T, Paavola P, Peltonen L Assignment of the locus for hydrolethalus syndrome to a highly restricted region on 11q23-25. Am. J. Hum. Genet. 65, 1086–1095 (1999).
Lynch SA Non-multifactorial neural tube defects. Am. J. Med. Genet. Part C, Semin. Med. Genet. 135, 69–76 (2005).
Bennett RL, French KS, Resta RG, Doyle DL Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J. Genet. Couns. 17, 424–433 (2008).
Acknowledgements
This work was supported by the Polish grant of Medical University of Bialystok UMB 123-06843 L, UMB 3 06 574 L, Polish Ministry of Science and Higher Education, Grant No N407 03432/1371, Grant NN301 238836, and National Science Centre in Poland, Grant 2011/01/B/NZ2/04819. We are grateful for dr Ryszard Lesniewicz and for dr Grazyna Sorbaj-Sucharska for participation in pedigree data collection and for Anna Sawicka for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Midro, A., Panasiuk, B., Stasiewicz-Jarocka, B. et al. Recurrence risks for different pregnancy outcomes and meiotic segregation analysis of spermatozoa in carriers of t(1;11)(p36.22;q12.2). J Hum Genet 59, 667–674 (2014). https://doi.org/10.1038/jhg.2014.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.92
This article is cited by
-
Chromosome (re)positioning in spermatozoa of fathers and sons – carriers of reciprocal chromosome translocation (RCT)
BMC Medical Genomics (2019)
-
Limited survivability of unbalanced progeny of carriers of a unique t(4;19)(p15.32;p13.3): a study in multiple generations
Molecular Cytogenetics (2017)
-
Unrevealed mosaicism in the next-generation sequencing era
Molecular Genetics and Genomics (2016)
-
Meiotic and pedigree segregation analyses in carriers of t(4;8)(p16;p23.1) differing in localization of breakpoint positions at 4p subband 4p16.3 and 4p16.1
Journal of Assisted Reproduction and Genetics (2016)
