
Abstract
Phenelfamycins G and H are new members of the family of elfamycin antibiotics with the basic structure of phenelfamycins E and F, respectively, which are also well known as ganefromycins α and β. Phenelfamycins G and H differ from phenelfamycins E and F by an additional hydroxy group at position C-30, which is not described so far for any of the elfamycin-type antibiotics. The actinomycete strain that produced phenelfamycins G and H was identified to be Streptomyces albospinus based on its 16S rRNA gene sequence. Phenelfamycins G and H exhibit a narrow antibacterial spectrum with a pronounced inhibitory activity against Propionibacterium acnes.
Similar content being viewed by others
Introduction
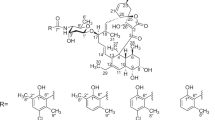
Environmental samples were collected from untapped Malaysian ecological niches to isolate actinomycetes strains, which were investigated within our HPLC-diode array screening program for the production of secondary metabolites with the aim of detecting novel drugs for pharmaceutical applications.1 Strain Acta 3619 was isolated from a soil collected from the rhizosphere of bamboo trees in the tropical rainforest of the University of Malaya Field Station at Gombak, Selangor, Malaysia. The strain was found to be of special interest, as it gave two significant peaks in the HPLC analysis of an extract of the culture filtrate having congruent UV–vis spectra that differed from all 937 reference compounds stored in our HPLC–UV–vis database.2 Structure elucidation of the two isolated compounds revealed a relationship with phenelfamycins E and F and therefore were named as phenelfamycins G (1) and H (2), respectively. Their structures are shown in Figure 1.

Structures of phenelfamycins G (1) and H (2).
Phenelfamycins belong to the elfamycin family of antibiotics, which inhibit protein synthesis of bacteria by an interaction with elongation factor Tu, demonstrated in prokaryotic cells.3, 4, 5 Hence they derived their name from this interaction. Elfamycins can be subdivided into three types according to their structure. Type 1 includes kirromycin6 (mocimycin), aurodox,7, 8 heneicomycin9 and efrotomycin.10 Type 2, elfamycin, is represented by kirrothricin with an open tetrahydrofuran ring in the aglycone.11 Finally, type 3 contains the phenelfamycins with a phenylacetyl substitution at position C-22 or C-23 and C-33 O-glycosylation with mono- (phenelfamycins A and B), di- (phenelfamycins C and D) or trisaccharide units (phenelfamycins E and F, also named as ganefromycins α and β).12, 13
All elfamycins show a very narrow antibacterial spectrum against human pathogens and most of them show excellent activities as growth-promoting agents in animals.14
In this paper we report on the new phenelfamycins 1 and 2, the taxonomy of the producing strain, their fermentation, isolation, structure elucidation and biological activity.
Results
Taxonomy of the producing strain
Strain Acta 3619 was isolated from the rhizosphere soil sample from a patch of bamboo at the University of Malaya Field Station in Gombak Selangor on the west coast of Malaysia (3°19′N and 101°45′E). Strain Acta 3619 grew on a wide range of media, except ISP5 and ISP7 (Table 1). Its cultural and phenotypic characteristics are given in Table 2. The sporulation was poor on all media studied except ISP2. The strain grew well in a temperature range of 27–37 °C and an NaCl concentration of 0–1%. The strain contained L,L-diaminopimelic acid in the cell wall. The scanning electron microscopic observation revealed that aerial mycelium on ISP2 media had a single turn of spiral spores and the spore surface was smooth (Figure 2). On the basis of growth and growth characteristics, strain Acta 3619 was assigned to the genus Streptomyces. The 16S rRNA gene sequence of the strain is 100% similar to Streptomyces albospinus NBRC 13846T (Accession No. AB184527) (Figure 3).

Micrograph of the aerial mycelium of strain Acta 3619 with a single turn of spore chain classified in Spira and Rectiflexibiles; scale bar: 1 μm.

Neighbor-joining tree based on nearly complete 16S rRNA gene sequences showing relationships between strain Acta 3619 and representatives of closely related Streptomyces species. The numbers at the nodes indicate the level of bootstrap support (%) based on the analysis; scale bar: substitutions per nucleotide position.
Screening, fermentation and isolation
Strain Acta 3619 was grown in 500 ml Erlenmeyer flasks in various complex media. Samples of 10 ml volume were taken between 3 and 6 days of incubation and extracts were prepared from both culture filtrates and mycelia and analyzed by HPLD-DAD. Two dominant peaks with retention times of 9.3 min (1) and 9.9 min (2) were detected in the culture filtrate extract when strain Acta 3619 was cultivated in oatmeal medium (Figure 4). Their identical UV–vis spectra differed significantly from those of the 937 reference compounds—mostly antibiotics—stored in our in-house-developed HPLC–UV–vis database. A search in the Dictionary of Natural Products15 gave no positive identification.

HPLC analysis of a culture filtrate extract from Streptomyces sp. Acta 3619 at a fermentation time of 96 h, monitored at 230 nm. 1=phenelfamycin G; 2=phenelfamycin H; insert: UV-Vis spectrum of 1 and 2, respectively.
A scale-up of the cultivation of strain Acta 3619 was done in the 20-l fermentor scale in oatmeal medium, in which maximal growth was reached at 72 h. The production of 1 started at 24 h and increased continuously till 72 h, when maximal production was reached with an amount of 45 mg l−1. The production of 2 was initialized at a later period and was observed at 96 h of incubation, reaching an amount of 12 mg l−1. Compounds 1 and 2 were isolated from the culture filtrate by Amberlite XAD-16 chromatography and subsequent extraction with ethyl acetate and chromatography on diol-modified silica gel and Sephadex LH-20. Both compounds were separated by preparative reversed-phase HPLC, yielding white powders after lyophilization.
Structure elucidation
The physico-chemical properties of phenelfamycins G (1) and H (2) are summarized in Table 3. The IR absorption spectrum contains a broad band around 3347 cm−1 covering the region for OH groups. The bands at 1728 and 1643 cm−1 indicate carboxylic acid and an amide group to be present in the molecule. Structure elucidation was carried out using the information obtained from MS and standard 2D NMR spectroscopy (COSY, TOCSY, ROESY, HSQC and HMBC). The ESI-MS spectra of phenelfamycin G (1) and H (2) showed molecular ions at m/z=1264.8 ([M+Na]+) and 1240.8 ([M–H]−). The exact molecular masses were determined by high-resolution ESI-MS as 1242.6410 ([M+H]+) for G (1) and 1242.6404 ([M+H]+) for H, (2) and gave the molecular formula of C65H95O22N for 1 and 2. After H/D exchange with D2O/CD3OD, the ESI-MS spectra of 1 and 2 showed molecular ions at m/z=1271.8 ([M(D)+Na]+) and 1246.8 ([M(D)-D]−), which led to seven exchangeable protons. The 13C NMR data, including DEPT/HSQC, showed 11 methyl groups, 8 methylenes, 39 methins and 7 quaternary carbons in both compounds. The 1D NMR spectra of 1 and 2 were very similar, with only small differences. Because of this similarity, we would like to present only the structure elucidation of 2 in detail and point out only the differences for 1.
Trisaccharide
Three doublets at δH=4.66, 4.78 and 4.90 corresponding to δC=98.3, 95.9 and 99.7 were identified as anomeric protons of the three sugars that are part of a trisaccharide, proved by HMBC correlations. Constitutions of the trisaccharide sugars A, B and C were determined from COSY and HMBC as 2,6-didesoxy-3-O-methyl-hexoses.
Sugar A is determined as α-sugar by the proton–proton coupling constant of J1′-H,2′-H=2.3 Hz and the proton–carbon coupling constant of 1JC-1′,1′-H=169.2 Hz. 3′-H shows an axial–axial coupling (J2′-Hax,3′-H=11.8 Hz) and is therefore in axial position. 4′-H shows no axial–axial coupling and is therefore in equatorial position. In the ROESY NMR spectrum, a crosspeak 3′-H/5′-H is visible and proves the axial position of 5′-H. Thus, sugar A is determined to be α-oleandose.16
Sugar C is an α-sugar determined from J1′″-H,2′″-H=3.2 Hz and proton–carbon coupling constant 1JC-1′″,1′″-H=168.8 Hz. 3′″-H shows an axial–axial coupling (J2′″-Hax,3′″-H=12.0 Hz) and is therefore in axial position. 4′″-H shows no axial–axial coupling and is therefore in equatorial position. In the ROESY NMR spectrum, a crosspeak 3′″-H/5′″-H is visible and proves the axial position of 5′″-H. Thus, sugar C is determined to be α-oleandose.16
The proton–proton coupling constant of J1″-H,2″-H=9.2 Hz and proton–carbon coupling constant (1JC-1″,1″-H=161.9 Hz) showed sugar B to be a β-sugar. Although sugar B has the same constitution like sugars A and C, it shows a different conformation. The conformation was determined from ROESY NMR experiment by crosspeaks 1″-H/5″-H and 4″-H/2″-Hax, which proves these protons to be axial, supported by the axial–axial coupling J4″-H,5″-H=9.7 Hz. Sugar B could be identified as β-cymarose.17
The linkage between the three sugars was given by the HMBC crosspeaks C-1″/4′-H, C-1′″/4″-H and C-4″/H-1′″ (Figure 5). Through strong ROESY couplings we were able to achieve the preferred conformation of the three sugar units within the trisaccharide by correlations 4′-H/1″-H, 4″-H/1′″-H, 6′-H/1″-H and 6″-H/1′″-H (Figure 5). The trisaccharide unit is α-oleandose–β-cymarose–α-oleandose and fits to the trisaccharide earlier described in phenelfamycins.12, 13

Selected 1H,1H-COSY, ROESY and HMBC correlations in the trisaccharide chain of 2.
The linkage between the trisaccharide chain and the aglycone is established through HMBC couplings between C-1′/33-H and C-33/1′-H (Figure 5).
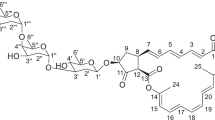
Tetrahydrofuran-bearing chain
The chain starts with the carboxylic group C-1 (δC=167.7), and the connectivity to the trien unit is established by HMBC signals C-1/2-H and C-1/3-H. The conjugated trien chain in the UV spectra leads to an absorption maximum at 235 nm. The configuration has been determined as E for the trien double bonds C-2/C-3, C-4/C-5, C-6/C-7 due to alternating coupling constants of J∼15.0 Hz (2-H/3-H, 4-H/5-H and 6-H/7-H) and ROESY correlations for 3-H/5-H, 5-H/7-H, 2-H/4-H and 6-H/4-H (Figure 6).

Selected 1H,1H-COSY, ROESY and HMBC correlations in the tetrahydrofuran bearing chain in 2.
The trien chain (C-1 to C-7) ends in a tetrahydrofuran unit with three stereogenic centers (C-8, C-9 and C-11), which is substituted by a hydroxyl group at C-9 (δC=73.3). We found strong ROESY signals between 8-H/6-H, 10-Ha/9-H, 10-Ha/8-H and 10-Hb/11-H, 10-Ha/31-H3, 10-Hb/12-H, which led to the following orientations: 8-H pseudoaxial, 9-H pseudoequatorial, 9-OH pseudoaxial, 10-Ha pseudoaxial, 10-Hb pseudoequatorial and 11-H pseudoaxial (Figure 7 I). In Newman projection, it can be illustrated that ROESY signals 31-H3/10a-H and 8-H/31-H3 are possible in case of pseudoequatorial orientation of C-12, so 11-H must be anti to the methyl group 31-H3 (Figure 7 II). The conformation between C-12 and C-13 can be monitored in Newman projection too. We found ROESY signals between 13-H/11-H, 13-H/31-H3, 13-H/13-OCH3 and to the adjacent olefinic unit with couplings between 13-H/32-H3 and 13-H/15-H. We did not observe ROESY correlations between 12-H and 13-H that support anti-orientation of the protons 13-H and 12-H. The confirmation of these anti-orientations required an additional 1D-TOCSY experiment (excitation of proton 11-H δH=4.46) that revealed H-13 (δH=3.32) as a well-separated doublet with a coupling constant of J13-H,12-H=9.5 Hz, which implicates proton 13-H to be anti to 12-H. The ROESY couplings between 13-H/32-H3 and 13-H/15-H are possible, through parallel orientation of the double bond C-14/C-15 to 31-H3 (Figure 7 III).

Conformation and configuration of the tetrahydrofuran moiety and the adjacent side chain (C-12–C-16) in 2.
The two double bonds C-14/C-15 and C-16/C-17 were identified to be in E configuration on the basis of ROESY crosspeaks 15-H/17-H and 16-H/32-H3 (Figure 6). The chemical shift of the methylene group C-18 (δC=40.7) fits perfectly to the adjacent amide group C-19-NH (δC=174.0). Furthermore, on the basis of the COSY signal 18-H/NH and the HMBC signal C-19/H-18, the connectivity of the amide group was established (Figure 6). The linkage of the tetrahydrofuran bearing chain to the trisaccharide is given through the HMBC signals C-19/20-H and C-19/33-H (Figure 6). The missing connectivity between the tetrahydrofuran-bearing chain and the hemiacetal in HMBC spectra measured in DMSO-d6 was monitored after running additional spectra in CDCl3 (Supplementary Tables S7 and S8 in Supporting Information). The HMBC spectra in CDCl3 revealed the important coupling C-21/H-20 (δC=98.4/δH=3.02), indicating the linkage to the hemiacetal.
Hemiacetal
The cyclic structure of the hemiacetal has been established from the HMBC signals C-21/22-H, C-21/25-H, C-23/H-22, C-23/H-25, C-24/H-22, C-24/H-23 and C-24/H-25 shown in Figure 8. The quaternary carbon C-24 is substituted by the two methyl groups indicated by HMBC signals C-25/34-H3 and C-25/35-H3, and from the two methyl groups C-34 and C-35 to the two methine groups 23-H and 25-H. The cyclic hemiacetal is substituted by a diene at C-25. The coupling constant J26-H,27-H=15.2 Hz established the double bond at C-26/C-27 as E supported by ROESY signals H-26/H-28 (Figure 8). The double bond between C-28/C-29 showed Z configuration, proved by the coupling constant J28-H,29-H=10.1 Hz and ROESY signals H-28/H-29 and H-27/H-30 (Figure 8). The chemical shift at C-30 (δC=57.0) indicated an adjacent OH group proved by an H/D-exchange experiment. The OH group is the decisive new element, which distinguishes phenelfamycins G (1) and H (2) from phenelfamycins E and F.

Structure determination of the hemiacetal in 2.
Like phenelfamycins E and F, G (1) and H (2) differ in their esterification at C-22-OH (1) or C-23-OH (2) through a phenylacetyl group, leading to significant differences in their chemical shifts, especially at C-22, C-23 and C-34 (Table 4). The esterification at C-23-O in phenelfamycin H (2) is monitored by the HMBC signals C-36/H-23. The ROESY signals H-23/H-25 and H-37/H-20 prove the equatorial orientation of the phenylacetyl group at C-23 (Figure 9). Further, a ROESY signal H-23/H-22 supports the axial orientation of the hydroxyl group at C-22 in 2.

Conformation in the hemiacetal unit of phenelfamycins G (1) and H (2).
The esterification at C-22-O in phenelfamycin G (1) is indicated by the HMBC signal C-36/H-22. The phenylacetyl group at C-22 (δC=71.5) has to be axial because of the ROESY signal H-37/H-34 and is supported by further ROESY signals H-25/H-35 and H-34/H-26, suggesting the axial orientation of H-34 (Figure 9).
Finally, the structures of phenelfamycins G and H are determined as 1 and 2 (Figure 1).
Biological activity
Phenelfamycins G (1) and H (2) were tested against Gram-positive and Gram-negative bacteria. Both compounds showed a weak activity against Xanthomonas campetris and a pronounced activity against Propionibacterium acnes (IC50=0.55±0.05 μM for 1 and IC50=0.80±0.10 μM for 2). Compounds 1 and 2 were found to be inactive against all other tested microorganisms and thus showed a narrow antibacterial spectrum comparable to other members of the elfamycin antibiotic family. Moreover, 1 and 2 showed weak inhibition of acetylcholine esterase. Acetylcholine esterase hydrolyzes the neurotransmitter acetylcholine and is a major therapeutic target for the symptomatic treatment of Alzheimer's disease.18 In addition, both compounds did not affect the other tested enzyme activities and did not inhibit cell proliferation.
Discussion
Strain Acta 3619 was identified as a strain similar to S. albospinus based on morphological and chemotaxonomic characteristics, as well as 16S rRNA analysis. It was reported that strain M 750–G1 producing spinamycin, a non-polyene antifungal compound active against Trichophyton sp., was related to S. albus.19 As the strain had spines on its spores, whereas spores of S. albus were smooth, the strain was named as S. albospinus. Another antifungal compound, phenamide was isolated from S. albospinus A19301 having a spiny spore ornamentation and 5–6 turns of spiral spores.20 In contrast, strain Acta 3619 has no spore surface ornamentation, although various physiological characteristics were similar to S. albospinus strains. Furthermore, molecular data based on 16S rRNA sequence revealed a 100% homology with S. albospinus. Thus, strain Acta 3619 was identified as a variant of S. albospinus and designated as ‘S. albospinus subsp. Albus’.
The two new phenelfamycins isolated from strain Acta 3619 are derivatives of phenelfamycin E (ganefromycin α) and phenelfamycin F (ganefromycin β) with an additional hydroxylation at position C-30 and named phenelfamycin G (1) and H (2). Hydroxylation at position C-30 is not described by other elfamycin antibiotics so far.
The determined NMR-data resemble those of elfamycin antibiotics and the relative configurations from proton–proton couplings and mainly ROESY couplings fit perfectly to those earlier reported for phenelfamycin E (ganefromycin α) and F (ganefromycin β).12, 21 As an evidence for the absolute configuration of phenelfamycin H (2) the CD spectrum is in good accordance to that of phenelfamycin E (ganefromycin α).21 The results from high-resolution mass spectra leading to molecular formula of C65H95NO22 and the H/D-exchange experiments confirming seven exchangeable protons support the presence of an additional OH group in contrast to phenelfamycin E (ganefromycin α) and F (ganefromycin β). Thus, phenelfamycins G (1) and H (2) can be described as novel analoges of phenelfamycin E and F, ganefromycin α and β, respectively, with an additional OH group at C-30 that was not found for other elfamcyin antibiotics yet.
From phenelfamycins E and F an excellent growth-promoting activity in animals was reported,12 showing similar inhibitory activities as clindamycin against gram-positive anaerobes, including Propionibacterium acnes and Clostridium difficile.22 As other phenelfamycin compounds, phenelfamycin G (1) and H (2) showed a pronounced activity against Propionibacterium acnes that led to the potential of a usage in cosmetics for treatment of acne.
Methods
General experiments
NMRspectra were measured on a VNMR-S 600 MHz spectrometer (Varian, Walnut Creek, CA, USA) equipped with 3-mm triple resonance inverse and 3-mm dual broadband probeheads. Spectra are recorded in 150 μl DMSO-d6 at T=35 °C. Solvent signals were used as internal standard (DMSO-d6: δH=2.50, δC=39.5). Proton–carbon coupling constants are measured via coupled HSQC.
UV-Vis spectra were measured on a Varian Cary 100 double-beam spectrophotometer equipped with 1.0-cm cuvettes.
HR-ESI mass spectra were performed on a LTQ Orbitrap XL (Thermo Fisher Scientific, Waltham, MA, USA).
HPLC-DAD-ESI-MS analysis was done with an Agilent 1200 HPLC series equipped with a binary HPLC pump, autosampler, diode array detector, and a LC/MSD Ultra Trap System XCT 6330 (Agilent, Waldbronn, Germany). A volume of 2.5 μl of the samples were injected onto an HPLC column (Nucleosil-100 C-18, 3 μm, 100 × 2 mm i.d.) and separated by 0.1% aqueous HCOOH as solvent A and 0.06% HCOOH in CH3CN as solvent B by a linear gradient from 10% B to 100% B over 15 min at a flow rate of 400 μl min−1.
The H/D-exchange mass experiment was performed using an Agilent LC/MSD Ultra Trap system XCT 6630 with a syringe pump. All parts were flushed with CD3OD/D2O/acetic acid-d4 before the experiment.
CD spectra are recorded on a Chirascan spectrometer (Applied Photophysics, Leatherhead, UK) equipped with 0.2 and 1.0 cm cuvettes.
FTIR spectra (spectral resolution of 4 cm−1, 1000 scans) were obtained using a Bruker IFS 66V FTIR spectrometer (Bruker, Ettlingen, Germany) equipped with a liquid nitrogen-cooled cryogenic mercury cadmium telluride detector. Background spectra were obtained using EtOH, CHCl3 cleaned silicon wafers. The samples were dropped as CDCl3 solution on the silicon wafer surface. The organic solvent was removed through a slight nitrogen stream to achieve a thin film of the sample.
Producing organism and taxonomy
Rhizosphere soil sample was collected from a bamboo clump at the University of Malaya Field Station at Gombak, Selangor Darul Ehsan. The soil sample was air-dried for 3 days before moist heat treatment.23 The sample was then diluted in 0.9% NaCl and 0.1 ml of the 10−3 diluted sample was plated on starch-casein agar incorporated with antifungal and antibacterial agents. The inoculated plates were then incubated at 27±2 °C and observed periodically for putative actinomycete strains.24 The axenic culture of strain Acta 3619 was transferred to yeast extract-malt extract agar (ISP2). Standard protocols were then used for the observation of growth characteristics and identification of the strain.25, 26 Color grouping was based on fluorescent light observation and categorization according to color codes of Meuthen Handbook of Colors.27 The culture grown on ISP2 media for 14 days was observed for aerial and substrate mycelium and spore chain morphology by scanning electron microscope (Phillips SEM 15; FEI Singapore). The culture was exposed to osmium tetroxide vapor for 4 h and then mounted on aluminum stubs before coating with gold.
The hydrolysate of whole cell of strain Acta 3619 grown on ISP2 medium was analyzed for diaminopimelic acid type.28 The carbohydrate usage pattern and physiological characteristics were studied based on standard protocols.25, 28
Genomic DNA extraction and PCR amplification of the 16S rRNA gene were carried out as described previously.29 The 16S rRNA gene sequence of the strain was aligned using ClustalW30 against corresponding sequences of representatives of closely related Streptomyces spp., as retrieved from the EzTaxon database.31 A neighbor-joining tree was inferred with the Jukes & Cantor (1969) algorithm and the topology of the resultant tree was evaluated by using a bootstrap analysis using the TREECON program.32
HPLC-diode array analysis
A 10-ml aliquot of the fermentation broth was centrifuged, and the supernatant adjusted to pH 5 and extracted with the same volume of EtOAc. After centrifugation, the organic layer was concentrated to dryness in vacuo and resuspended in 0.5 ml MeOH. Aliquots (10 μl) of the samples were injected onto an HPLC column (125 × 4.6 mm i.d.) fitted with a guard-column (20 × 4.6 mm i.d.) filled with 5-μm Nucleosil-100 C-18 (Maisch, Ammerbuch, Germany). The samples were analyzed by linear gradient elution using 0.1% ortho-phosphoric acid as solvent A and CH3CN as solvent B at a flow rate of 2 ml min−1. The gradient was from 0 to 100% for solvent B in 15 min with a 2-min hold at 100% for solvent B. The chromatographic system consisted of a HP 1090M liquid chromatograph equipped with a diode-array detector and a HP Kayak XM 600 ChemStation (Agilent). Multiple-wavelength monitoring was performed at 210, 230, 260, 280, 310, 360, 435 and 500 nm, and UV–vis spectra measured from 200 to 600 nm.
Fermentation and isolation
Batch fermentations of strain Acta 3619 were carried out in a 20-l fermentor equipped with a turbine impeller system (b20; B. Braun, Melsungen, Germany) in a complex medium that consisted of (per liter tap water) oatmeal (Holo Hafergold, Neuform; Zarrentin, Germany) 20 g, and trace element solution 5 ml, which was composed of (per liter deionized water) CaCl2·2H2O 3 g, iron(III) citrate 1 g, MnSO4·H2O 200 mg, ZnCl2 100 mg, CuSO4·5H2O 25 mg, Na2B4O7·10H2O 20 mg, CoCl2·6H2O 4 mg and Na2MoO4·2H2O 10 mg; the pH was adjusted to 7.3 (5 M HCl) before sterilization. The fermentor was inoculated with 5% by volume of a shake flask culture grown in a seed medium at 27 °C in 500 ml Erlenmeyer flasks with a single baffle for 72 h on a rotary shaker at 120 r.p.m. The seed medium consisted of glucose 10 g, glycerol 10 g, oatmeal 5 g, soybean meal (Schoenenberger, Magstadt, Germany) 10 g, yeast extract (Ohly Kat; Deutsche Hefewerke, Hamburg, Germany) 5 g, Bacto casamino acids 5 g and CaCO3 1 g in 1-l tap water. The fermentation was carried out for 4 days with an aeration rate of 0.5 volume air per volume and minute, and agitation at 1000 r.p.m.
Hyflo Super-cel (2%, Johns-Manville, Denver, CO, USA) was added to the fermentation broth, which was separated by multiple sheet filtration into culture filtrate and mycelium. The culture filtrate (15 l) was applied to an Amberlite XAD-16 column (resin volume 1.5 l, Rohm and Haas, Frankfurt, Germany) and the resin was washed with H2O and H2O-MeOH (1:1). 1 and 2 were eluted with 100% MeOH and concentrated in vacuo to an aqueous residue (600 ml). The concentrate was adjusted to pH 4.0 and extracted three times with EtOAc (each 120 ml), and the organic extracts combined and concentrated in vacuo to dryness. The crude product was dissolved in CH2Cl2 and applied to a diol-modified silica gel column (40 × 2.6 cm i.d.; LiChroprep Diol, Merck, Darmstadt, Germany). 1 and 2 were purified by step gradient elution using CH2Cl2 and MeOH as solvents at a flow rate of 420 ml h−1, starting with CH2Cl2 and were eluted by a MeOH content of 5%. Further purification was achieved by chromatography on Sephadex LH-20 (90 × 2.5 cm i.d.) with MeOH at a flow rate of 30 ml h−1. 1 and 2 were separated by preparative reversed phase-HPLC column (Reprosil-Pur Basic C18, 10 μm, 25 × 2.0 cm i.d.; Maisch, Ammerbuch, Germany) and linear gradient elution with 0.1% HCOOH as solvent A and CH3CN as solvent B. The gradient was from 45% B to 60% B in 15 min at a flow-rate of 20 ml min−1, yielding 48 mg and 32.4 mg of pure compound 1 and 2, respectively.
Antimicrobial assays
Antimicrobial assays were performed using Bacillus subtilis DSM 347, Staphylococcus epidermidis DSM 20044, Staphylococcus lentus DSM 6672, Erwinia amylovora DSM 50901, Escherichia coli K12 DSM 498, Pseudomonas fluorescens NCIMB 10586, Propionibacterium acnes DSM 1897, Pseudomonas aeruginosa DSM 50071, Pseudomonas syringae pv. aptata DSM 50252, Ralstonia solanacearum DSM 9544, Xanthomonas campestris DSM 2405 and the Candida glabrata DSM 6425. Overnight cultures of the test organisms in tryptic soy broth were diluted to an OD600 nm of 0.02–0.06. The assays were prepared by transferring 2 μl of a 10 mM solution of the sample compound dissolved in DMSO into one well of a 96-well microtiter plate containing 200 μl of cell suspension culture. After the microtiter plates were incubated for 14–16 h at 28 °C, 10 μl of a resazurin solution (0.2 mg ml−1 phosphate-buffered saline) was added to each well and the plates were incubated at 28 °C for 1–2 h. To evaluate cell viability, the reduction of resazurin to resorufin was assessed by measuring the intensity of fluorescence at 560Ex/590Em nm. The resulting values were compared with a positive (100 μM chloramphenicol for bacteria; 100 μM cycloheximide for the yeast) and a negative control (no compound) on the same plate. Propionibacterium acnes was grown anaerobically (Anaerocult A mini; Merck, Darmstadt, Germany) in peptone-yeast extract-glucose (PYG)-medium (modified DSMZ-medium 104) at 37 °C for 24–48 h. The bacterial culture was diluted to an OD600 nm of 0.03, 200 μl of the inoculum were added to each well and the microtiter plate was incubated anaerobically at 37 °C for 48 h.
Cytotoxicity assays
The sensitivity of the cell lines NIH-3T3 and HepG2 to the isolated compounds was evaluated by monitoring the metabolic activity using the CellTiter-BlueCell Viability Assay (Promega, Mannheim, Germany). The mouse fibroblast cell line was kindly provided by G. Rimbach (University of Kiel, Germany). The human hepatocellular carcinoma cell line HepG2 was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). HepG2 and NIH-3T3 cells were maintained in RPMI 1640 medium. Media were supplemented with 10% fetal bovine serum (Promocell, Heidelberg, Germany), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Invitrogen, Carlsbad, CA, USA). The cultures were maintained at 37 °C under a humidified atmosphere and 5% CO2. The cell lines were transferred every 3 or 4 days. For experimental procedures, cells were seeded in 96-well plates at concentrations of 7500 cells (NIH-3T3) and 10 000 cells (HepG2) per well. After 24-h incubation, the medium was removed and 100 μl of the test sample adjusted to final concentrations of 10 μM by diluting in growth medium was added to the cells. Each sample was prepared in triplicate. Tamoxifen, as a standard therapeutic drug was used as a positive control. Following compound addition, plates were cultured for 24 h at 37 °C. Afterwards, the assay was performed according to the manufacturer's instructions. Cells were incubated for 2 h at 37 °C. Fluorescence was measured using the microplate reader Infinite M200 (Tecan, Männedorf, Switzerland) at excitation 560 nm and emission 590 nm.
Enzyme activity assays
To uncover specific enzyme inhibitory activities, the isolated compounds were screened in several enzyme activity tests including prominent drug targets such as phosphodiesterase 4 (PDE4), protein tyrosine phosphatase N1 (PTPN1), HIV-1 reverse transcriptase (HIV-1-RT) and acetylcholine esterase (AchE). AchE activity was measured using adaptations of the colorimetric assay described by Ellman et al. for a microplate test system.33 Putative inhibitory activities against human recombinant protein tyrosin phosphatase N1 (PTP1B) were tested using the Biomol Green PTP1B tyrosin phosphatase drug discovery kit (catalog number AK822-0001, Biomol, Hamburg, Germany). Reverse transcriptase activity was assayed using a colorimetric reverse transcriptase enzyme-linked immunosorbent assay (ELISA) kit (catalog number 11468120910, Roche, Mannheim, Germany) according to the manufacturer's instructions. The activity of phosphodiesterase 4 (PDE4) was measured using the PDE Light HTS cAMP phosphodiesterase kit (catalog number LT07-600, Lonza, Rockland, ME, USA) according to the manufacturer's instructions.
References
Gebhardt, K., Meyer, S. W., Schinko, J., Bringmann, G., Zeeck, A. & Fiedler, H.-P. Phenalinolactones A-D, terpenoglycoside antibiotics from Streptomyces sp. Tü 6071. J. Antibiot. (e-pub ahead of print 22 December 2010; doi:10.1038/ja.2010.165).
Fiedler, H- P. Biosyntheticcapacities of actinomycetes. 1. Screening for novel secondary metabolites by HPLC and UV-visible absorbance libraries. Nat Prod. Lett. 2, 119–128 (1993).
Wolf, H., Chinali, G. & Parmeggiani, A. Kirromycin, an inhibitor of protein synthesis that acts on elongation factor Tu. Proc. Nat Acad. Sci. USA 71, 4910–4914 (1974).
Hall, C. C., Watkins, J. D. & Georgopapadakou, N. H. Effects of elfamycins on elongation factor Tu from Escherichia coli and Staphylococcus aureus. Antimicrob. Agents Chemother. 33, 322–325 (1989).
Vogeley, L., Palm, G. J., Mesters, J. R. & Hilgenfeld, R. Conformational change of elongation factor Tu (EF-Tu) induced by antibiotic binding: Crystal structure of the complex between EF-Tu.GDP and aurodox. J. Biol. Chem. 276, 17149–17155 (2001).
Wolf, H. & Zähner, H. Stoffwechselprodukte von Mikroorganismen, 99. Mitteilung. Kirromycin. Arch. Mikrobiol. 83, 147–154 (1972).
Berger, J., Lehr, H. H., Teitel, S., Maehr, H. & Grunberg, E. A new antibiotic X-5108 of Streptomyces origin. I. Production, isolation and properties. J. Antibiot. 26, 15–22 (1973).
Maehr, H., Leach, M., Williams, T. H. & Blount, J. F. The chemistry of aurodox and related antibiotics. Can. J. Chem. 58, 501–526 (1980).
Dewey, R. S., Hensens, O. D., Douglas, A. W. & Albers-Schönberg, G. The structure of heneicomycin. J. Antibiot. 44, 838–843 (1991).
Wax, R., Maiese, W., Weston, R. & Birnbaum, J. Efrotomycin, a new antibiotic from Streptomyces lactamdurans. J. Antibiot. 29, 670–673 (1976).
Thein-Schranner, I., Zähner, H., Hoppe, H. U., Hummel, I. & Zeeck, A. Kirrothricin, a new member of the kirromycin-group. J. Antibiot. 35, 948–956 (1982).
Hochlowski, J. E. et al. Phenelfamycins, a novel complex of elfamycin-type antibiotics. J. Antibiot. 41, 1300–1315 (1988).
Carter, G. T., Phillipson, D. W., West, R. R. & Borders, D. B. Chemistry and structure of ganefromycin. J. Org. Chem. 58, 6588–6595 (1993).
Maise, W. M. et al. LL-E19020α and β, animal growth promoting antibiotics: taxonomy, fermentation and biological activity. J. Antibiot. 42, 1489–1493 (1989).
Dictionary of Natural Products on DVD. Version 18:2, (CRC Press, London, UK 2009).
Pauli, G. F. Adoligoses, oligosaccharides of rare sugars from Adonis aleppica. J. Nat. Prod. 58, 483–494 (1995).
Kumar, A., Khare, A. & Kare, N. K. Trisaccharides from Marsdenia roylei. Phytochemistry 52, 675–679 (1999).
Melnikova, I. Therapies for Alzheimer's disease. Nature Rev. Drug Discovery 6, 341–342 (2007).
Wang, E. L., Hamada, M., Okami, Y. & Umezawa, H. A new antibiotic, spinamycin. J. Antibiot. 19, 216–221 (1966).
Makkar, N. S. et al. Phenamide, a fungicidal metabolite from Streptomyces albospinus A19301 - taxonomy, fermentation, isolation, physico-chemical and biological properties. J. Antibiot. 48, 369–374 (1995).
Andersson, T., Berova, N., Nakanishi, K. & Carter, G. T. Relative and absolute configurations of ganefromycin α. Org. Lett. 2, 919–922 (2000).
Swanson, R. N. et al. Phenelfamycins, a novel complex of elfamycin-type antibiotics. J. Antibiot. 42, 94–101 (1989).
Schneider, K. et al. Nocardichelins A and B, a new group of siderophores from Nocardia sp. Acta 3026. J. Nat. Prod. 70, 932–935 (2007).
Helaly, S. et al. Gombapyrones, new—pyrone metabolites produced by Streptomyces griseoruber Acta 3662. J. Antibiot. 62, 445–452 (2009).
Shirley, E. B. & Gottlieb, D. Methods for characterisation of Streptomyces species. Int. J. Syst. Bacteriol. 16, 313–340 (1966).
Takashi, S. et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. J. Antibiot. 57, 173–179 (2004).
Kornerup, A. & Wanscher, J. H. Meuthen Handbook of Colors (1961)-Color Codes 2nd edn (Meuthen, London, UK, 1967).
Lechavelier, M. P. & Lechevalier, H. The chemotaxonomy of actinomycetes in Actinomycetes Taxonomy Vol. 6 (eds Dietz, A. & Thayer, D. W.) 227–291 (Society of Industrial Microbiology, Fairfax, 1980).
Tan, G. Y. A., Ward, A. C. & Goodfellow, M. Exploration of Amycolatopsis diversity in soil using genus-specific primers and novel selective media. Syst. Appl. Microbiol. 29, 557–569 (2006).
Larkin, M. A. et al. ClustalW and ClustalX version 2. Bioinformatics 23, 2947–2948 (2007).
Chun, J. et al. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 57, 2259–2261 (2007).
Van de Peer, Y. & De Wachter, R. TREECON for Windows: a software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput. Applic. Biosci. 10, 569–570 (1994).
Eliman, G. L., Courtney, K. D., Andres, V. Jr & Featherstone, R. M A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 (1961).
Acknowledgements
Financial support from the European Commission (project ACTAPHARM, 5th framework, QLK3-CT-2001-01783) and the Siegener Studienstiftung is gratefully acknowledged. We thank Mr J Nachtigall, Technical University of Berlin for HRMS analysis and Mr R Halip, Institute of Biological Sciences, University of Malaya for the SEM analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Article no. 58 in ‘Biosynthetic Capacities of Actinomycetes’. Article no. 57: see reference 1. On the occasion of Prof. Harald Günthers' 75th birthday.
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Brötz, E., Kulik, A., Vikineswary, S. et al. Phenelfamycins G and H, new elfamycin-type antibiotics produced by Streptomyces albospinus Acta 3619. J Antibiot 64, 257–266 (2011). https://doi.org/10.1038/ja.2010.170
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.170
Keywords
This article is cited by
-
Pharmacological Potential of Phylogenetically Diverse Actinobacteria Isolated from Deep-Sea Coral Ecosystems of the Submarine Avilés Canyon in the Cantabrian Sea
Microbial Ecology (2017)
-
Langkocyclines: novel angucycline antibiotics from Streptomyces sp. Acta 3034
The Journal of Antibiotics (2013)
-
Benzoxacystol, a benzoxazine-type enzyme inhibitor from the deep-sea strain Streptomyces sp. NTK 935
The Journal of Antibiotics (2011)
