
Abstract
Purpose:
Mowat–Wilson syndrome (MWS) is a genetic disease characterized by distinctive facial features, moderate to severe intellectual disability, and congenital malformations, including Hirschsprung disease, genital and eye anomalies, and congenital heart defects, caused by haploinsufficiency of the ZEB2 gene. To date, no characteristic pattern of brain dysmorphology in MWS has been defined.
Methods:
Through brain magnetic resonance imaging (MRI) analysis, we delineated a neuroimaging phenotype in 54 MWS patients with a proven ZEB2 defect, compared it with the features identified in a thorough review of published cases, and evaluated genotype–phenotype correlations.
Results:
Ninety-six percent of patients had abnormal MRI results. The most common features were anomalies of corpus callosum (79.6% of cases), hippocampal abnormalities (77.8%), enlargement of cerebral ventricles (68.5%), and white matter abnormalities (reduction of thickness 40.7%, localized signal alterations 22.2%). Other consistent findings were large basal ganglia, cortical, and cerebellar malformations. Most features were underrepresented in the literature. We also found ZEB2 variations leading to synthesis of a defective protein to be favorable for psychomotor development and some epilepsy features but also associated with corpus callosum agenesis.
Conclusion:
This study delineated the spectrum of brain anomalies in MWS and provided new insights into the role of ZEB2 in neurodevelopment.
Genet Med advance online publication 10 November 2016
Similar content being viewed by others

Introduction
Mowat–Wilson syndrome (MWS; OMIM 235730) is caused by heterozygous deletions or loss-of-function variants of the ZEB2 gene, which maps on region 2q22.3 (GRCh38 genomic coordinates 2:144,384,374-144,520,390). Clinical manifestations include large, deep-set eyes; hypertelorism; large medially flaring and sparse eyebrows; rounded nasal tip; prominent columella; M-shaped upper lip; pointed triangular chin; uplifted, fleshy ear lobes with a central depression; and microcephaly. Additional features are moderate to severe intellectual disability, epilepsy, Hirschsprung disease, hypospadias in males, congenital heart disease, brain anomalies, and eye defects. The basic genomic defect is heterogeneous, including 2q21–q23 deletions encompassing ZEB2 and additional contiguous genes or partial gene deletions in a minority of subjects and intragenic loss-of-function variants, such as frameshift, stop gain, and splice-site variants, in most. Missense variants rarely occur in association with a typical MWS phenotype. Since the first description by Mowat in 1998, and after the definition of the distinct MWS phenotype with or without Hirschsprung disease, more than 270 cases of a proven ZEB2 haploinsufficiency have been reported so far.1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16
ZEB2 (zinc finger E-box binding homeobox 2), also known as ZFHX1B (zinc finger homeobox 1B) or SIP1 (Smad-interacting protein 1),17 is a member of the ZEB family of zinc finger transcription factors, which are essential during normal embryonic development. ZEB family members induce epithelial to mesenchymal transition (EMT), a process that reorganizes epithelial cells to become migratory mesenchymal cells and is essential for developmental processes such as gastrulation, neural crest formation, heart morphogenesis, formation of the musculoskeletal system, and craniofacial structures18,19,20,21 (Supplementary Figure S1 and Supplementary Table S1 online). In human embryos, ZEB2 was found to be homogeneously expressed throughout the central nervous system (CNS), from mesencephalon to spinal cord.19
Of note, a mouse model expressing a conditional mutation in the Zeb2 gene in neural cell precursor cells clearly indicated a crucial role of Zeb2 for proper neural crest cell development.22 Cranial neural crest cells differentiate into connective tissue and skeletal–muscular structures of the head, whereas vagal neural crest cells reach the gastrointestinal tract and the cardiovascular system. Therefore, nearly all MWS-associated clinical signs can be explained by a defect in the induction, migration, and differentiation of neural crest cells, thus fitting the description of a neurocristopathy.
Consistently, neurological involvement is a main feature of MWS in humans. Moderate to severe intellectual disability is present in all patients; epilepsy has a prevalence of 70–75% and has an age-dependent electroclinical pattern.8,14,23 Since the delineation of this disorder in 1998 by Mowat and Wilson, there has been an increasing appreciation of distinctive structural and functional neurologic anomalies in this condition. Previous studies point to several recurrent brain abnormalities in MWS. The most frequent are agenesis of corpus callosum (ACC) and corpus callosum hypoplasia (HCC), which account for 46% of the published cases.5,14,24 According to the definition by Edwards et al.,25 we consider “ACC” to encompass both total absence (complete ACC) and absence of at least one, but not all, of the anatomically defined regions of the corpus callosum (partial ACC), which results in a shorter anterior–posterior length. Hypoplasia denotes a corpus callosum that is thinner than usual but has a normal anterior–posterior extent. However, in the majority of papers, the authors do not specify what they meant by the terms “partial ACC” and “hypoplasia of corpus callosum.” It is likely that some of the patients reported to have hypoplasia had partial ACC, or vice versa, and that therefore the frequency of this finding has been misrepresented.
Other reported brain anomalies are ventriculomegaly,11 cortical atrophy,6 pachygyria and cerebellar hypoplasia,26 and poor hippocampal formation.3,7 Although apparently rare, these findings are more likely to be underrepresented because no extensive neuroimaging studies involving individuals with MWS have been conducted so far—not all published cases involved cranial imaging, and some clinical case reports failed to mention any magnetic resonance imaging (MRI) studies. In particular, no literature studies have taken into account the comparison of the neuroimaging data with direct examination of brain MRI in a wide population of MWS patients.
Here, we performed a systematic longitudinal evaluation of brain MRI studies involving 54 MWS patients with the aims of further characterizing the spectrum of brain abnormalities in this syndrome, investigating genotype–phenotype correlations, and gaining new insights into the role of ZEB2 in human brain development.
Materials and Methods
Cohort analysis
A cohort of 54 patients (23 males and 31 females) with molecularly confirmed ZEB2 haploinsufficiency and available brain MRI data was assembled through the collaborative efforts of the contributing authors. Genetic and molecular studies are summarized in Supplementary Table S2 online. All cases were sporadic except for two affected sisters (subjects 29 and 30).
Each patient underwent head MRI from 1 to 12 separate times, for a total of 74 neuroimaging studies ( Table 1 ). Minimum requirements for MRI were (i) three-dimensional sagittal T1-weighted (T1-w) images with coronal, axial, and sagittal reconstructions; (ii) axial and coronal T2-weighted (T2-w) images; and (iii) axial fluid attenuation inversion recovery (FLAIR) images.
A direct and systematic longitudinal review of the complete MRI data from the original CD-ROMs of all 74 studies was performed by two multidisciplinary teams consisting of neuroradiologists, pediatricians, pediatric neurologists, and medical geneticists. In addition to assessment for novel features, findings from a literature review were used as a checklist for evaluating the presence or absence of specific features reported in MWS ( Table 1 and Supplementary Table S3 online).
Types of epileptic seizures and psychomotor development were assessed via both direct examination of the patients (neurological, electroencephalography, MRI) and evaluation of their medical records.
All procedures involving patients performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Written informed consent was obtained.
Evaluation of neuroimaging findings in the literature
In our review of the literature, encompassing all the brain neuroimaging findings reported so far, we identified 109 cases of both brain MRI described and MWS confirmed by ZEB2 gene analysis. For proper comparison with our cohort, we considered only the 56 cases for which it was clearly described that MRI had been evaluated completely and in detail (Supplementary Table S3 online).
Statistical analysis
Characteristics of patients’ neuroimaging findings, epilepsy, and psychomotor development were analyzed by means of descriptive statistics. Differences in the listed categorical characteristics between patients with predicted synthesis or absence of the variant protein were assessed using the Fisher exact test with P < 0.05 as the level of significance.
Results
Neuroimaging features
In our cohort, only 2 of 54 patients (3.7%) had normal neuroimaging findings, whereas 25 of 56 (44.6%) in the literature had normal MRI findings ( Figures 1 and 2 , and Supplementary Figure S2 online); 83.3% of our patients had from 2 to 7 abnormalities, with an average of 3.54.

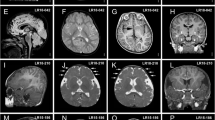
Brain magnetic resonance imaging findings in the cohort and in the literature.

Brain magnetic resonance imaging findings in the cohort. (a) Complete agenesis of corpus callosum; (b) partial agenesis of corpus callosum; (c) hypoplasia of corpus callosum; (d) hippocampal abnormalities; (e) lateral ventricle enlargement and reduction of white matter thickness; (f) localized signal alteration of white matter; (g) cortical dysplasia; (h) ventricular temporal horn enlargement; (i) large basal ganglia; (j) posterior fossa findings; and (k) periventricular nodular heterotopia.
A distinctive feature observed in more than half of the patients was a commissural defect. Corpus callosum abnormalities were documented for most patients and consisted of complete ACC, partial ACC (localized in the caudal or in the rostral third), or hypoplasia with thin corpus callosum.
We made a comparison between the corpus callosum abnormalities in our study and the 56 in the literature that had both MRI described in detail and MWS confirmed by ZEB2 gene analysis.
In our series, 14 patients (25.9%) had complete agenesis of corpus callosum compared with 23.2% of the literature. In our group, partial agenesis of corpus callosum was observed in nine patients (16.67%) compared with 1.8% of the literature. In the majority of the cases the agenesis was typical, with frontal residue, splenium absence, and preservation of the genu.
MRI showed hypoplasia of the corpus callosum with thin corpus callosum in 20 patients (37.04%), whereas it was found in 23.2% of those in the literature.
White matter abnormalities were commonly observed findings, including reduction of thickness, which was widespread in the whole brain or more evident in one or more lobes. Reduction of white matter thickness was present in 22 patients (40.7%) compared with 17.9% in the literature. Localized signal alterations of white matter were present in 22.2% of our cases (12 patients); comparison with data in the literature was not possible because insufficient information was available.
Concurrently, ventriculomegaly was present, with widening of the temporal or occipital horns. Lateral ventricle enlargement was evident in 38.9% of our cases (21 patients) compared with 12.5% in the literature, and ventricular temporal horn enlargement was detected in 59.26% of our series (32 patients) compared with 12.5% in the literature.
Bilateral hippocampal abnormalities were noted in more than half of our population (42 patients, 77.8%) with morphological or positional anomalies, whereas in the literature these abnormalities were noted in only 7.1% of cases. Large basal ganglia, a rare anomaly not previously reported in MWS, was observed in 3 patients (5.6%).
Other findings included two cases of hypoplastic cerebellum and one case each of polymicrogyria,27 periventricular heterotopia, focal cortical dysplasia, Chiari type 1 malformation, absent cerebellar vermis, small cerebellar vermis, and macrocerebellum. We also examined one case of CNS tumor, which had already been reported previously.28
Genotype–phenotype correlations with MRI findings
The types of mutation in our cohort included large deletions (in eight patients: n = 8), small deletions/insertions/indels with frameshift (n = 21), and nonsense mutations (n = 25). According to protein synthesis prediction (taking into account the position of premature stop codons and possible mRNA degradation29), all of these variants can be stratified into three major groups: (A) complete deletion of a ZEB2 allele (n = 7), (B) mutations predicted to lead to absence of the protein product (n = 40), and (C) mutations predicted to lead to synthesis of a defective protein (n = 7).
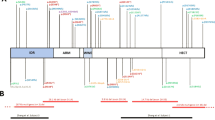
To allow statistical analysis, we also considered individuals with ZEB2 whole-allele deletion, patients with other variants that lead to absence of the protein in a single group (A+B; n = 47), and patients with predicted synthesis of a defective protein in another group (C; n = 7). Based on these categories, we delineated a genotype–phenotype correlation for the neurological features found in our patients ( Figure 3 ).

Genotype–phenotype correlation. (a) Brain magnetic resonance imaging features; (b) seizure onset; (c) types of seizures and resistance to therapy; and (d) developmental milestones.
Overall, anomalies of the corpus callosum were very frequent in all groups: 87.5% (n = 6/7) in group A, 72.5% (n = 29/40) in group B, and 100% (n = 7/7) in group C ( Figure 3a ). However, some heterogeneity emerged by observing specific defects: complete ACC was most frequent in group C (71.4%, n = 6/7), whereas it was rare in group B (12.5%, n = 5/40) and fairly common in group A (57.1%, n = 4/7). When considering all variants leading to predicted protein absence, we found a statistically significant difference in the occurrence of complete ACC (19.14% in group A+B vs. 71.4% in group C; P = 0.0095).
Hippocampal abnormalities were present in at least 71.4% individuals in each group, with a frequency of 100% in group C ( Figure 3a ).
Genotype–phenotype correlations with epilepsy
Epilepsy is one of the main features of MWS. In our cohort, 91.8% of the patients (n = 45/49) had seizures, with a mean onset at 29.7 months.
The earliest onset was at age 3 months in a patient from group A; the latest onset was in a patient at age 10 years. Patients in group A generally had an earlier onset, with a mean of 24.4 months, whereas patients in groups B and C had average onsets at 32 and 30 months, respectively ( Figure 3b ). Four patients in our cohort—all in group B—have not manifested seizures to date: two of them are younger than 2 years, one is 4 years old, and the oldest is 11.5 years old.
Fever-triggered seizures occurred in 55.8% of patients (n = 24/43), with the highest frequency in group C (66.6%, n = 4/6) and the lowest in group A (16.67%, n = 1/6).
Focal, absence, and generalized tonic clonic seizures were equally represented in all three groups (79.3–83.3, 50–60, and 43.85–60% respectively).
Focal seizures with secondary generalization were more frequent in group B (51.8%, n = 14/27); in groups A and C they occurred in 40% (n = 2/5) and 16.67% (n = 1/6) of cases, respectively. None of the patients with complete deletion of a ZEB2 allele presented with myoclonic seizures, whereas in group B and group C the frequencies were 24 and 33.3%, respectively. None of the patients in group C had resistance to antiepileptic drugs, whereas in groups A and B epilepsy was resistant in 50% (n = 3/6) and 29.6% (n = 8/27) of cases, respectively ( Figure 3c ).
Genotype–phenotype correlations with psychomotor development
All patients with typical MWS display psychomotor developmental delay, evaluated on the basis of sitting age, walking age, and age of first words. Based on our results, patients with predicted synthesis of a defective protein (group C) have the least severe delay of all three groups.
Considering only the cases for which we have valid data, mean ages of sitting without support were 17.14 months in group C (n = 7/7), 19.98 in group B (n = 26/40), and 24 in group A (n = 4/7). Mean ages of autonomous walking were 44.67, 49.78, and 80 months in groups C, B, and A, respectively. Speech was the most severely affected feature for all patients with MWS. In our cohort, the mean age of first words was 40.5 months in group C, 43.2 months in group B, and undetermined in group A (the exact onset of speech was available only for one 9-year-old patient). At least 60% of the patients with complete deletion of ZEB2 had complete absence of speech ( Figure 3d ); the rest of the patients were capable of saying a few words (fewer than five).
Discussion
MWS could be conceived as an in vivo model of the consequences of ZEB2 haploinsufficiency on human neurodevelopment, and neuroimaging is an important tool for highlighting its effects on the macrostructural anatomy of the CNS. To our knowledge, this study is the first attempt to carefully examine the neuroradiological characteristics of MWS in a large cohort of patients and in light of recent advances concerning the role of ZEB2 in neuroembryology.
Through systematic brain MRI analysis, we delineated a neuroradiological phenotype of MWS, characterized by a high frequency of complete or partial ACC, hypoplasia of corpus callosum (thin corpus callosum), hippocampal abnormalities (particularly malrotation), reduction of white matter thickness, and anomalies in the cerebral ventricles such as dilatation and temporal horn enlargement. Less common but relevant findings were large basal ganglia, polymicrogyria, nodular periventricular heterotopia, cortical dysplasia, cerebellar hypoplasia, Chiari type 1 malformation, and localized signal alteration of white matter ( Figures 1 and 2 , and Table 1 ).
Previous studies showed a stable rate of 46% of corpus callosum anomalies (ACC) across different populations.5,14,24 Our study highlights that commissural defects are present, at least partially, in a much greater proportion of cases. In addition to complete ACC, several grades of partial ACC and HCC are often displayed, and the other commissures can also be altered in patients with MWS. In particular, partial ACC was almost 10 times more frequent than in previously reported cases.
Other structural brain abnormalities were very common in our cohort, with most patients displaying more than one relevant feature, but were rarely reported in previously published works. In particular, hippocampal anomalies and enlargement of ventricles were, respectively, 10 and 6 times more common in our cohort. Some other features, such as localized signal alterations of the white matter and large basal ganglia, have not been previously described for MWS. Our results suggest that all these may be underrecognized findings.
Alterations of the corpus callosum in MWS are consistent with a mouse model described by Srivatsa et al.,30 which indicated that Zeb2 is essential for the formation of intracortical, intercortical, and corticosubcortical connections in the murine forebrain: Zeb2-deficient neocortical neurons fail to form the corpus callosum, anterior commissure, and corticospinal tract.
In another mouse model, Weng et al.31 demonstrated that Zeb2 acts as a master regulator for myelination in the CNS, connecting extracellular signaling pathways to intracellular transcriptional programs and promoting the transition from immature to mature myelinating oligodendrocytes. This would explain the reduction in thickness or signaling alterations of the white matter consistently observed in 40.7 and 22.2% of our patients, respectively.
Hippocampal morphological and/or positional abnormalities were found in more than half of our population. This could be the consequence of the commissural defects, but direct involvement of ZEB2 cannot be excluded. In fact, Miquelajauregui highlighted the role of Zeb2 in the development of the hippocampus, as its inactivation causes death of differentiating cells and decreased proliferation in the region of the prospective hippocampus in mutant mice.32 Analysis by Nishizaki confirmed its expression in the pyramidal hippocampal neurons.33
Two patients showed cerebellar hypoplasia, which has been reported.26 We speculate a possible role of ZEB2 in this malformation because Nishizaki demonstrated the expression of Zeb2 in Bergmann cells in the cerebellum of mice.33
Furthermore, brain anatomy does not seem to discriminate the cognitive outcome among the analyzed patients, who show a marked, global impairment.
Genotype–phenotype correlations
One aim of this study was to delineate a genotype–phenotype correlation for the neurological features of MWS. More than 100 different variations have been described in the literature in patients with clinically typical MWS. They almost always consist of whole-gene deletions or intragenic truncating mutations (nonsense or frameshift) of ZEB2, suggesting that haploinsufficiency is the basis of MWS pathology. No obvious genotype–phenotype correlation has been identified to date,12 but atypical phenotypes have been reported in association with missense or specific splice-site mutations in the ZEB2 gene, indicating that some sort of genotype–phenotype correlation in MWS probably exists.34,35
In the absence of a detailed functional characterization of each ZEB2 variant, we compared our neurological findings (brain MRI features, EEG and epilepsy features, psychomotor developmental milestones) with those for three broad genotypic categories determined by the predicted effect of the genomic variation on protein synthesis.
Because some literature reports suggest a more severe phenotype in association with large locus deletions, we separated those cases into a group separate from predicted protein absence due to intragenic mutations.
In addition, a few reported variants are expected—or, in rare cases, have been proven—to affect the protein product in other ways: missense mutations leading to defective repressor activity or protein processing or splice-site mutations eliminating specific functional domains or favoring a documented alternative splicing.8,12,34,35,36 However, their numbers are extremely small: less than 5% of all reported cases worldwide and less than 2% if we consider only typical MWS. None of these were present in our cohort.
Most brain MRI features have no significant relationship to these genotypic groups. Overall, corpus callosum anomalies are equally represented in all categories; however, when observing specific defects, complete ACC seems particularly infrequent in patients with protein absence due to intragenic mutations ( Figure 3a ). By considering together all variations with predicted absence of protein, variations expected to lead to a defective protein result significantly associated with complete ACC (P = 0.0095). Because most of the variants from group C are predicted to be truncated and likely nonfunctional proteins, we hypothesize that corpus callosum formation might be particularly sensitive to a dominant negative effect from protein accumulation or to some undefined feedback mechanism triggered by ZEB2 synthesis and lack of repressor activity.
Epilepsy
All patients with epilepsy had seizures regardless of the genotypic variation, but we observed some variability, depending on the type of seizure ( Figure 3b ,c). Although the numbers were small and could be used only for a descriptive analysis, patients with whole-allele deletion were recurrently associated with worsening features, particularly an earlier onset of seizures and a high incidence of drug resistance. However, the later onset of seizures and the lack of resistance to antiepileptic drugs indicate a slightly more favorable evolution of epilepsy in patients predicted to have synthesis of a defective protein.
The four patients who did not present with seizures were all predicted to have protein absence, but we would be cautious to assume that this might be due to simple statistical chance because group B is larger by far than the other two, leading to no significant correlation.
Epilepsy is often caused by malformations of cortical development, but these were rarely observed in our cohort. Furthermore, we found no evidence of significant correlation between the brain malformation phenotype, including hippocampal alterations, and the epilepsy phenotype in our patients. Even when cortical malformations were evident with brain MRI, no differences were observed in the characteristics of epilepsy because these patients showed the typical electroclinical pattern in patients with MWS.23
These observations, together with the recurrent clinical pattern and age-dependent development, reinforce the plausible hypothesis that epilepsy in MWS has a genetic etiology. ZEB2 has been demonstrated to play a crucial role in promoting cerebrocortical lamination37 and myelination in mice and humans, through antagonism of inhibitory signaling.31 Later studies have supported the influence of ZEB2 on the neurogenesis of cortical GABA-ergic interneurons: conditional deletion of Zeb2 in the medial ganglionic eminence of mice prevents the repression of the Nkx2-1 homeobox transcription factor, causing differentiation into striatal interneurons rather than cortical ones.38 This switch could explain the occurrence of epilepsy in MWS, as proposed by McKinsey, Cordelli, and van den Berghe and colleagues.38,39,40
Psychomotor development
All patients with typical MWS display psychomotor developmental delay. Based on our results, patients with predicted synthesis of a defective protein have the least severe delays of all three groups (sitting age, walking age, and first words age) ( Figure 3d ). The more severe developmental delay observed in patients with whole-ZEB2-allele deletion may correlate with the severity and type of epileptic seizures.
Conclusions
Several types of malformations can be detected with brain MRI, usually those involving the corpus callosum, hippocampus, and ventricular system. No cortical macrostructural abnormality appears to be clearly related to epilepsy onset and its features, which are probably genetically induced. CNS defects in MWS appear to correlate with the data observed in mice models and confirm in vivo the ZEB2 ability of regulating (at the least) myelination and hippocampus development. Our data are also consistent with ZEB2-expression patterns in the CNS of human embryos.19 Thus, the effects of ZEB2 haploinsufficiency in the diencephalon would explain the ACC observed in most patients and its defects in the mesencephalon could provide the basis for epilepsy, whereas its defects in the rhombencephalon could correlate with the severe intellectual disability.
We also found an indication that the effect of specific mutations could influence the clinical phenotype of MWS. Overall, our results suggest that the most favorable type of mutation for psychomotor development and for some aspects of epilepsy, such as drug-resistant epilepsy, would be that in which there is synthesis of a defective protein. However, this genomic category seems to be associated with some other features, such as complete ACC.
Specific functional studies are required to understand the biological properties of ZEB2 in MWS and clarify the molecular pathway of its protein–protein interactions, in order to facilitate prediction of the severity of this syndrome and enable the development of targeted therapeutic strategies.
Until then, the detailed brain MRI phenotype of MWS we provide here will help improve clinical suspicion and therefore the detection rate of this condition.
Disclosure
The authors declare no conflict of interest.
References
Mowat DR, Croaker GD, Cass DT, et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet 1998;35:617–623.
Amiel J, Espinosa-Parrilla Y, Steffann J, et al. Large-scale deletions and SMADIP1 truncating mutations in syndromic Hirschsprung disease with involvement of midline structures. Am J Hum Genet 2001;69:1370–1377.
Cacheux V, Dastot-Le Moal F, Kääriäinen H, et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet 2001;10:1503–1510.
Wakamatsu N, Yamada Y, Yamada K, et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat Genet 2001;27:369–370.
Zweier C, Albrecht B, Mitulla B, et al. “Mowat-Wilson” syndrome with and without Hirschsprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am J Med Genet 2002;108:177–181.
Garavelli L, Donadio A, Zanacca C, et al. Hirschsprung disease, mental retardation, characteristic facial features, and mutation in the gene ZFHX1B (SIP1): confirmation of the Mowat-Wilson syndrome. Am J Med Genet A 2003;116A:385–388.
Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet 2003;40:305–310.
Wilson M, Mowat D, Dastot-Le Moal F, et al. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am J Med Genet A 2003;119A:257–265.
Garavelli L, Cerruti-Mainardi P, Virdis R, et al. Genitourinary anomalies in Mowat-Wilson syndrome with deletion/mutation in the zinc finger homeo box 1B gene (ZFHX1B). Report of three Italian cases with hypospadias and review. Horm Res 2005;63:187–192.
Zweier C, Thiel CT, Dufke A, et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet 2005;48:97–111.
Adam MP, Schelley S, Gallagher R, et al. Clinical features and management issues in Mowat-Wilson syndrome. Am J Med Genet A 2006;140:2730–2741.
Dastot-Le Moal F, Wilson M, Mowat D, Collot N, Niel F, Goossens M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat 2007;28:313–321.
Garavelli L, Mainardi PC. Mowat-Wilson syndrome. Orphanet J Rare Dis 2007;2:42.
Garavelli L, Zollino M, Mainardi PC, et al. Mowat-Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet A 2009;149A:417–426.
Evans E, Einfeld S, Mowat D, Taffe J, Tonge B, Wilson M. The behavioral phenotype of Mowat-Wilson syndrome. Am J Med Genet A 2012;158A:358–366.
Wenger TL, Harr M, Ricciardi S, et al. CHARGE-like presentation, craniosynostosis and mild Mowat-Wilson Syndrome diagnosed by recognition of the distinctive facial gestalt in a cohort of 28 new cases. Am J Med Genet A 2014;164A:2557–2566.
Zollino M, Garavelli L, Rauch A. Clinical utility gene card for: Mowat-Wilson syndrome. Eur J Hum Genet 2011;19:.
Postigo AA, Dean DC. Differential expression and function of members of the zfh-1 family of zinc finger/homeodomain repressors. Proc Natl Acad Sci USA 2000;97:6391–6396.
Espinosa-Parrilla Y, Amiel J, Augé J, et al. Expression of the SMADIP1 gene during early human development. Mech Dev 2002;114:187–191.
Das S, Becker BN, Hoffmann FM, Mertz JE. Complete reversal of epithelial to mesenchymal transition requires inhibition of both ZEB expression and the Rho pathway. BMC Cell Biol 2009;10:94.
Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci 2009;66:773–787.
Van de Putte T, Francis A, Nelles L, van Grunsven LA, Huylebroeck D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum Mol Genet 2007;16:1423–1436.
Cordelli DM, Garavelli L, Savasta S, et al. Epilepsy in Mowat-Wilson syndrome: delineation of the electroclinical phenotype. Am J Med Genet A 2013;161A:273–284.
Yamada Y, Nomura N, Yamada K, et al. The spectrum of ZEB2 mutations causing the Mowat-Wilson syndrome in Japanese populations. Am J Med Genet A 2014;164A:1899–1908.
Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014;137(Pt 6):1579–1613.
Silengo M, Ferrero GB, Wakamatsu N. Pachygyria and cerebellar hypoplasia in a patient with a 2q22-q23 deletion that includes the ZFHX1B gene. Am J Med Genet A 2004;127A:109.
Murray SB, Spangler BB, Helm BM, Vergano SS. Polymicrogyria in a 10-month-old boy with Mowat-Wilson syndrome. Am J Med Genet A 2015;167A:2402–2405.
Valera ET, Ferraz ST, Brassesco MS, et al. Mowat-Wilson syndrome: the first report of an association with central nervous system tumors. Childs Nerv Syst 2013;29:2151–2155.
Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998;23:198–199.
Srivatsa S, Parthasarathy S, Molnár Z, Tarabykin V. Sip1 downstream Effector ninein controls neocortical axonal growth, ipsilateral branching, and microtubule growth and stability. Neuron 2015;85:998–1012.
Weng QJ, Chen Y, Wang HB, et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012;73:713–728.
Miquelajauregui A, Van de Putte T, Polyakov A, et al. Smad-interacting protein-1 (Zfhx1b) acts upstream of Wnt signaling in the mouse hippocampus and controls its formation. Proc Natl Acad Sci USA 2007;104:12919–12924.
Nishizaki Y, Takagi T, Matsui F, Higashi Y. SIP1 expression patterns in brain investigated by generating a SIP1-EGFP reporter knock-in mouse. Genesis 2014;52:56–67.
Ghoumid J, Drevillon L, Alavi-Naini SM, et al. ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum Mol Genet 2013;22:2652–2661.
Zweier C, Horn D, Kraus C, Rauch A. Atypical ZFHX1B mutation associated with a mild Mowat-Wilson syndrome phenotype. Am J Med Genet A 2006;140:869–872.
Balasubramaniam S, Keng WT, Ngu LH, Michel LG, Irina G. Mowat-Wilson syndrome: the first two Malaysian cases. Singapore Med J 2010;51:e54–e57.
Seuntjens E, Nityanandam A, Miquelajauregui A, et al. Sip1 (Zfhx1b) regulates sequential fate decisions through feedback signaling from postmitotic neurons to progenitor cells. Mech Develop. 2009;126:S202–S202.
McKinsey GL, Lindtner S, Trzcinski B, et al. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 2013;77:83–98.
Cordelli DM, Pellicciari A, Kiriazopulos D, Franzoni E, Garavelli L. Epilepsy in Mowat-Wilson syndrome: is it a matter of GABA? Epilepsia 2013;54:1331–1332.
van den Berghe V, Stappers E, Vandesande B, et al. Directed migration of cortical interneurons depends on the cell-autonomous action of Sip1. Neuron 2013;77:70–82.
Acknowledgements
The financial support of Telethon Italy (grant GEP 14131) is gratefully acknowledged. A.R.P. is supported by the National Institute of Neurological Disorders and Stroke under award K08NS078054.
The authors thank the Galliera Genetic Bank, member of the Telethon Genetic Biobank Network (project GTB12001), funded by Telethon Italy, and the Associazione Italiana Mowat Wilson ONLUS (AIMW), for assistance in managing patients’ samples. We also thank all cooperating family members for contributing the medical and neuroimaging data necessary for this study. In addition, we are grateful to Luca Valcavi for helping with the design and adaptation of the figures and tables.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figures and Tables
(ZIP 3148 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Garavelli, L., Ivanovski, I., Caraffi, S. et al. Neuroimaging findings in Mowat–Wilson syndrome: a study of 54 patients. Genet Med 19, 691–700 (2017). https://doi.org/10.1038/gim.2016.176
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.176
Keywords
This article is cited by
-
Mowat-Wilson syndrome: unraveling the complexities of diagnosis, treatment, and symptom management
Egyptian Journal of Medical Human Genetics (2024)
-
Identification of the DNA methylation signature of Mowat-Wilson syndrome
European Journal of Human Genetics (2024)
-
Brain size reductions associated with endothelin B receptor mutation, a cause of Hirschsprung’s disease
BMC Neuroscience (2021)
-
Zeb2 is a negative regulator of midbrain dopaminergic axon growth and target innervation
Scientific Reports (2017)
-
The alliance between genetic biobanks and patient organisations: the experience of the telethon network of genetic biobanks
Orphanet Journal of Rare Diseases (2016)
