
Abstract
Genome-wide studies of patients carrying pathogenic variants (mutations) in BRCA1 or BRCA2 have reported strong associations between single-nucleotide polymorphisms (SNPs) and cancer risk. To conduct the first genome-wide association analysis of copy-number variants (CNVs) with breast or ovarian cancer risk in a cohort of 2500 BRCA1 pathogenic variant carriers, CNV discovery was performed using multiple calling algorithms and Illumina 610k SNP array data from a previously published genome-wide association study. Our analysis, which focused on functionally disruptive genomic deletions overlapping gene regions, identified a number of loci associated with risk of breast or ovarian cancer for BRCA1 pathogenic variant carriers. Despite only including putative deletions called by at least two or more algorithms, detection of selected CNVs by ancillary molecular technologies only confirmed 40% of predicted common (>1% allele frequency) variants. These include four loci that were associated (unadjusted P<0.05) with breast cancer (GTF2H2, ZNF385B, NAALADL2 and PSG5), and two loci associated with ovarian cancer (CYP2A7 and OR2A1). An interesting finding from this study was an association of a validated CNV deletion at the CYP2A7 locus (19q13.2) with decreased ovarian cancer risk (relative risk=0.50, P=0.007). Genomic analysis found this deletion coincides with a region displaying strong regulatory potential in ovarian tissue, but not in breast epithelial cells. This study highlighted the need to verify CNVs in vitro, but also provides evidence that experimentally validated CNVs (with plausible biological consequences) can modify risk of breast or ovarian cancer in BRCA1 pathogenic variant carriers.
Similar content being viewed by others


Introduction
Carriers of BRCA1 pathogenic variants are at increased risk for developing breast cancer and/or ovarian cancer, but the precise level of these risks is uncertain. Estimates of the cumulative risks of breast and ovarian cancer by age 70 years for BRCA1 pathogenic variant carriers range from 44% to 75% and 43 to 76%, respectively.1 Studies exploring the cause for the range in risk estimates have provided evidence that genetic factors have a key role in modifying cancer risks for carriers.2 The Consortium of Investigators of Modifiers of BRCA1/BRCA2 (CIMBA) has facilitated a number of large studies, which have identified variants mapping to >20 loci that are associated with altered risk of breast or ovarian cancer in BRCA1 pathogenic variant carriers.3, 4, 5, 6 The effect size associated with each variant identified to date has been relatively small (hazard ratio<1.5), and together they account for only a fraction of heritable variation in risk in BRCA1 pathogenic variant-positive families.
Copy-number variants (CNVs) are estimated to cover 5–10%7 of the human genome, which is an order of magnitude greater than the number of base pairs (bp; ~15 Mbp; dbSNP Human Build 142) encompassed by the more commonly studied single-nucleotide polymorphisms (SNPs). Thus, based on base pair coverage, CNVs are responsible for the majority of genetic variability in human populations. CNVs have also been shown to partially overlap or fully encompass genes or regulatory sequences resulting in a range of biological changes, such as altered gene expression.8 Importantly, these inherited structural variants have a role in many complex diseases,9 and comprise a proportion of the mutation spectrum for known cancer syndromes, such as hereditary breast–ovarian cancer syndrome, Lynch syndrome and Li–Fraumeni syndrome.10 Moreover, recent genome-wide CNV studies have reported associations between a common deletion polymorphism overlapping APOBEC3 and risk of both breast and ovarian cancer.11, 12, 13 Thus, other common and rare CNVs may similarly affect genes involved in cancer-related pathways. The contribution of germline CNVs to variable risk in individuals with deleterious BRCA1 pathogenic variants is unknown.
In this paper, we conducted a large genome-wide CNV analysis of 2500 BRCA1 pathogenic variant carriers, with or without breast and/or ovarian cancer, using a previously published SNP-based genome-wide association study.14 To maximize the sensitivity for CNV discovery, multiple CNV calling algorithms were applied to the data set. Analyses identified several putative CNVs overlapping gene regions associated with risk of breast or ovarian cancer for BRCA1 pathogenic variant carriers and a requirement for validation in larger studies.
Materials and methods
Study population
A total of 2500 BRCA1 pathogenic variant carriers was drawn from 20 centers from North America, Europe and Australia as reported previously.14 Eligibility criteria for study participants included the following: (1) female carriers of pathogenic BRCA1 variants; (2) at least 18 years of age at recruitment; and (3) Caucasian self-reported ancestry. BRCA1 pathogenic variant carriers selected for the study were stratified into two groups consisting of women diagnosed with invasive breast cancer when younger than 40-years old (n=1250) and women who had not developed breast cancer or who had developed a first ovarian cancer when 35 years of age or older (n=1250). All BRCA1 pathogenic variants are listed in Supplementary Table S3 and deposited in the ClinVar database (Submission ID - SUB1994380; http://www.ncbi.nlm.nih.gov/clinvar/). All carriers were recruited for research studies using ethically approved protocols at host institutions.
CNV detection and quality control
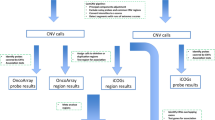
All DNA samples were genotyped with the Human610-Quad BeadChip (Illumina, Inc, San Diego, CA, USA) with ~610 000 markers (including ~20 000 non-polymorphic markers) for SNP and CNV analysis. Data for each array were normalized using GenomeStudio 2011.1 software (Illumina). Probe information including, genomic location, signal intensity (Norm R), allele frequency (Norm theta), Log R Ratios (LRRs), B allele frequencies (BAFs) for each sample was calculated and exported from GenomeStudio.
CNV calls were generated using four algorithms: PennCNV (version 2009 Aug27),15 QuantiSNP (v2.1),16 CNVPartition (v2.3.4, Illumina Inc.) and GNOSIS (a CNV detection algorithm within the CNV analysis package, CNVision, (http://sourceforge.net/projects/cnvision/files/). Quality control procedures were performed to remove poor quality array data (Supplementary Figure S3). Samples were excluded if they met the following criteria: PennCNV measures of log R ratio s.d.>0.28, BAF drift >0.01, waviness factor deviating from 0 by >0.05; QuantiSNP measures of BAF outliers >0.1, LogR outliers ≥0.1, BAF s.d. ≥0.2, LogR s.d. ≥0.4. A total of 2319 samples passed quality control steps and were assessed in the study. CNV calling results of all four algorithms were parsed and then merged using CNVision. To reduce false positives, CNV calls were excluded if ≥1000 kb in size, and/or were predicted by only one algorithm. Nine further CNVs called within the multi-histocompatibility complex on chromosome 6 were excluded from the study, as both a deletion and a duplication were predicted by two algorithms.
Defining CNV regions that may contribute to modification of risk
To identify new genomic loci contributing to breast or ovarian cancer risk in BRCA1 pathogenic variant carriers, common and rare deletions that overlapped gene regions were assessed using a genome-wide approach. Our study focused primarily on genomic deletions that overlapped gene regions for several reasons: (1) inter-individual analysis of CNVs is not straightforward as these variants do not typically occur in discrete genomic regions. The start and end coordinates of gene sequences were therefore used as a non-redundant approach to define CNV regions across the genome. (2) In contrast to duplications or copy-number gains, the genomic location of a deletion can be predicted from the array data. These data were not able to show the genomic location of a duplicated region, thus gene(s) or other functional genomic regions that are potentially disrupted by these structural events remain undetermined. (3) Whole or partial gene deletions are known to be potentially disruptive by causing haploinsufficiency or truncation of the expressed protein, and (4) common and rare CNVs that have previously been reported to be associated with breast and ovarian cancer risk have typically been deletion events.11, 12, 17, 18
We annotated 39 544 UCSC RefSeq (NCBI36/Hg18) transcripts using the SOURCE database19 and defined the genomic intervals for a total of 18 791 unique genes (Supplementary Figure S4). Thus, each gene interval encompassed the start and end of all corresponding alternate transcripts. CNVs and gene regions that were estimated to overlap by at least 1 bp were identified in a genome-wide scan using Intersect and Join tools from the Galaxy web server.20, 21, 22 All CNVs used for this study are deposited in the dbVar database (https://www.ncbi.nlm.nih.gov/dbvar) with the accession number nstd132.
CNV validation
Accessible DNA samples from the study cohort were used to validate 29 putative deletion regions. All predicted common (>1% frequency) deletions found associated with breast or ovarian cancer risk were chosen for validation. Copy-number assessment was carried out using Nanostring nCounter Elements TagSets (NanoString Technologies, Inc.) and Taqman assays. Target-specific Nanostring probes for 10 CNV and 10 invariant genomic regions are listed in Supplementary Table S1. Twenty-two gene regions were assessed using Taqman assay, including one region (GTF2H2) also analysed by with a Nanostring TagSet. Custom primer and probe sequences are presented in Supplementary Table S2. For seven CNV loci, we used the pre-designed assays from Life Technologies (Supplementary Table S2).
Statistical analysis
For the breast cancer risk association analysis, study participants (Supplementary Table S3) were classified at the age of the first breast cancer diagnosis or censored at ovarian cancer diagnosis or bilateral prophylactic mastectomy, whichever occurred first, or at the age of last observation. Only those diagnosed with breast cancer were considered as affected (n=1202 affected, n=1117 non-affected). Pathogenic variant carriers censored at their ovarian cancer diagnosis were considered to be unaffected in the breast cancer risk analyses. For ovarian cancer risk (n=357 affected, n=1962 non-affected), study participants were classified at the age of ovarian cancer diagnosis or censored at bilateral prophylactic oophorectomy, or age at last observation. Pathogenic variant carriers diagnosed with breast cancer were treated as unaffected at the age at breast cancer diagnosis. Analyses were carried out within a survival analysis framework. As BRCA1 pathogenic variant carriers were not randomly sampled with respect to their disease status, analyses were based on the modeling the retrospective likelihood of observing the CNV conditional on the observed phenotype.23 Two separate models were fitted to evaluate associations between CNVs with breast cancer and ovarian cancer risk, and were assessed using the 1 d.f. score test statistic.23 Q-values for the discrete test statistics were calculated by filtering the P-values using the T-method24 with a critical threshold of 0.05, such that genes with total number of deletions of four or more were retained.
Results
Genome-wide CNV analysis was performed on 2319 individuals with pathogenic BRCA1 pathogenic variants, including 1202 breast cancer cases (1117 non-breast cancer affected) and 357 ovarian cancer cases (1962 non-ovarian cancer affected), using published genotype data from Illumina 610K SNP arrays.14 A total of 60 893 CNVs were called across the study participants using four different algorithms (PennCNV, QuantiSNP, GNOSIS and CNVPartition) that passed the data quality threshold (see Methods and Materials). Of these, 89% and 94% CNVs were predicted by PennCNV and QuantiSNP, respectively, compared with a lower prediction rate from GNOSIS (35%) and CNVPartition (42%; Supplementary Table S4). The average number of CNVs observed per individual was 26.3 (range 4–203) that ranged in size from 314 to 999 990 bases.
A total of 21 013 CNVs were predicted to overlap at least one of 5848 different RefSeq genes across the study cohort. The average number of CNVs overlapping genes per individual was 9.1 (range 1–107). Deletions overlapping genes were detected approximately three times as often than duplications (6.8 versus 2.2, respectively). Interrogating the CNV calls from at least two algorithms revealed a deletion overlapping the BRCA1 gene in 14 study participants (Supplementary Figure S1). In each case, the deletion was confirmed by agreement with the results from the diagnostic BRCA1 germline genetic tests, supporting the use of two or more algorithms to reduce the possibility of artifactual CNV calling and false discoveries. However, CNV calling was unable to identify BRCA1 deletions overlapping five or more probes in eight pathogenic variant carriers that had previously been identified by diagnostic testing. These results therefore show a 100% detection specificity and a 64% detection sensitivity for CNV calls across the BRCA1 gene region. Two algorithms (PennCNV and QuantiSNP) dominated the CNV calling in this region, with PennCNV alone detecting a deletion in 14 cases (Supplementary Figure S1). No further BRCA1 deletions were identified using just one algorithm (data not shown).
Analysis of 5848 putative deletions delineated by gene regions identified a total of 52 loci associated (at unadjusted P<0.05) with breast cancer risk (Supplementary Table S6), and 72 CNV loci associated with ovarian cancer risk for BRCA1 pathogenic variant carriers (Supplementary Table S6). The top predicted CNV regions associated with risk included FGFR1OP2 (RR=0.20, P=5 × 10−4) and PABPC4L (RR=0.22, P=0.006) for breast and ovarian cancer, respectively. Eight loci (PABPC4L, APBA2, FAM189A1, FUT7, ENTPD2, NPDC1, C9orf139 and L1CAM) were associated with risks for both breast cancer and ovarian cancer (P<0.05).
SNP arrays are well known for low accuracy when assessing CNVs, compared with other platforms such as bacterial artificial chromosome array and oligonucleotide arrays.25 We therefore attempted to validate CNV regions using Nanostring technology, qPCR and data from the recently published Human CNV Map.7 Twenty-nine predicted CNV loci were selected for validation including the most common deletions (>1% frequency) found to be associated with breast or ovarian cancer risk in the BRCA1 pathogenic variant carrier cohort. Eight of these 29 (28%) CNV loci were confirmed by qPCR and/or Nanostring analysis, including four loci that were associated with breast cancer (GTF2H2, ZNF385B, NAALADL2 and PSG5) and two loci that were associated with ovarian cancer (CYP2A7 and OR2A1; Table 1). Nanostring analysis of eight putative CNV loci from Table 1 (ZNF385B, CALCRL, TFPI, GTF2H2, SLCO1B1 FGFR1OP2, TM7SF3 and ALX1) in 48 study samples only found seven deletions not identified by the calling algorithms, suggesting a low false negative rate (2% (7Nanostring calls/352Negative bioinformatic calls); Supplementary Table S5). The strongest association with a validated deletion was observed for ovarian cancer, detected in 75/1962 (3.8%) unaffected carriers and 4/357 (1.1%) affected carriers (RR=0.50, P=7 × 10−3) overlapping the CYP2A7 locus (19q13.2; Supplementary Figure S2).
To assess the functional relevance of the validated CNV deletion overlapping the CYP2A7 locus, the genomic landscape at this region was investigated using publicly available genomic data from ENCODE26 and the Roadmap Epigenomics Consortium27 (Figure 1). Examining data generated from normal ovarian tissue, the CNV deletion coincided with enhancer-specific histone modifications (acetylation of H3 lysine 27 (H3K27Ac) and mono-methylation of H3 lysine 4 (H3K4Me1)) and DNaseI hypersensitivity sites representative of open chromatin. By contrast, there was no evidence for these chromatin features in normal breast epithelial (HMEC) cells. Cross-reference to super-enhancers annotated in the study by Hnisz et al,28 found the CNV deletion overlapped an enhancer, found in ovary tissue, predicted to affect the expression of EGLN2, located ~67 kb downstream of CYP2A7.

Genomic landscape at the region containing the CNV deletion overlapping CYP2A7. The location of the CNV deletion and the enhancer predicted by Hnisz et al28 to affect EGLN2 are shown by black bars. Histone modifications associated with enhancer elements (H3K27Ac and H3K4Me1) and DNAseI hypersensitivity sites (HSs) for normal ovarian tissue and normal breast epithelial cells (HMECs) from Roadmap Epigenomics Consortium and ENCODE are depicted by histogram tracks.
Zarrei et al7 recently published a Human CNV Map constructed from multiple studies in the Database of Genomic Variants by applying a clustering algorithm to define ~27 000 CNV regions with high stringency. Comparing this stringent map with validated CNVs from this study revealed a strong consensus. All eight CNV loci validated in BRCA1 pathogenic variant carriers were present in the published CNV Map, and only one CNV (CNTNAP3B) that was not verified in our data was present in the CNV Map (Table 1). Using the published Human CNV Map to support the existence of putative CNVs from this association study identified deletions at nine of 52 gene loci (17%) that are associated with breast cancer risk (Supplementary Table S6), and 13 of 72 (18%) gene loci associated with ovarian cancer risk (Supplementary Table S7). With the exception of the CYP2A7 locus (P=0.007), all validated CNV regions returned a modest association (P-values ranged from 0.01 to 0.049) for ovarian or breast cancer risk. Validated CNVs ranged in allele frequency from 0.2 to 7.8%.
Discussion
Compared with SNPs, the contribution of CNVs to genetic variability and breast and/or ovarian cancer risk is relatively unknown. This is the first genome-wide CNV association study of BRCA1 pathogenic variant carriers to identify CNVs that are associated with breast and/or ovarian cancer risk, and the first implementation of the retrospective likelihood to CNV data. Our study used multiple CNV calling algorithms with the aim of increasing the sensitivity and specificity of CNV detection. Initial assessment of known deletions overlapping the BRCA1 gene indicated 100% detection specificity and 64% detection sensitivity. This assessment also showed that all 14 CNVs identified at BRCA1 were called by two or more calling algorithms, setting the calling criteria for the remainder of the study. However, validation of 29 predicted CNVs throughout the genome confirmed <30% of predicted deletions, highlighting a large number of false variant calls. None of the nine rare variants (<1% allele frequency) chosen for validation was verified by qPCR or Nanostring. However, CNV calling correctly predicted 40% (8/20) deletions we tested which ranged in allele frequency from 1.2 (OR2A1 locus) to 8.2% (ZNF385B locus). These results confirm other published reports that indicate array-based CNV data can be unreliable without further validation using ancillary technologies, such as qPCR.25 The accuracy may be increased by employing more stringent criteria but likely at the expense of detection sensitivity. For example, a larger number of probe markers could be used to generate a CNV call, but this approach will also reduce the spatial resolution of the array and sensitivity. PennCNV algorithm called ~90% of variants assessed in this study including all the deletions that were detected across BRCA1 in 14 cases. These data suggest that the combination of four algorithms for generating putative CNV information may not have been a vast improvement over using PennCNV alone.
Our study focused on genomic deletions that overlapped gene regions, as this approach provided functionally important genomic regions for comparing CNV calls. A notable finding was an association of a CNV deletion at the CYP2A7 locus (19q13.2) with decreased ovarian cancer risk (RR=0.50, P=0.007). To our knowledge, this locus has not previously been associated with cancer risk from SNP-based or CNV-based genome-wide association studies and requires further investigation. CYP2A7 encodes a member of the cytochrome P450 superfamily of enzymes, although the substrate(s) for this gene have not yet been determined. The deletion variant in this region may also affect the regulation of a nearby gene CYP2A6,29 which is known to have a key role in the metabolism of a number of substrates including nicotine, coumarin and valproic acid.30 Interestingly, a deletion at the CYP2A6 locus has been found to be associated with decreased risk of lung cancer in Asian smokers,31 which is comparable to our finding that CYP2A7 deletions were more frequent in non-affected high-risk BRCA1 pathogenic variant carriers compared with those with ovarian cancer (MAF—3.8% versus 1.1%). Examining published data from The Cancer Genome Atlas showed that ~40% of high-grade serous ovarian tumors, including 6% BRCA1 pathogenic variant carriers, exhibited somatic hemizygous deletions overlapping CYP2A7.32 Moreover, these deletions correlated with a reduced expression level compared with copy neutral CY2A7 (Supplementary Figure S5). These data indicate that, although a germline deletion of CYP2A7 may protect against initiation of ovarian cancer in the context of a BRCA1 germline pathogenic variant, somatic deletions of CYP2A7 may be important for the ovarian cancer development or progression.
Analysis of chromatin features from normal ovary tissue at the CYP2A7 genomic region shows that the CNV deletion coincides with chromatin marks consistent with an enhancer element. Interestingly, there was no evidence of similar features in normal breast epithelial cells, suggesting a tissue-specific feature. These results are concordant with the association of this CNV deletion with ovarian, and not breast, cancer risk in this cohort. Cross-reference of this region to the catalog of enhancers compiled by Hnisz et al28 found the CNV deletion overlaps a putative enhancer in ovarian tissue. This enhancer is predicted to affect expression of EGLN2, which encodes an enzyme involved in oxygen homeostasis. Further biological experiments are required to delineate the mechanism underlying the observed association between the CNV deletion and ovarian cancer risk. Importantly, although we prioritized CNVs for analysis based on overlap with coding genes, our findings suggest that intergenic CNVs could confer risk by altering regulatory elements. Therefore, future analyses integrating chromatin features into the CNV selection process could identify other CNVs, missed in this analysis, associated with cancer risk.
Confirmed deletions overlapping a total of nine gene loci were found associated with breast cancer risk, and a total of 13 gene loci associated with ovarian cancer risk in BRCA1 pathogenic variant carriers (Supplementary Table S5). GTF2H2 (5q13.2) is a transcription factor with a role in the nucleotide excision repair (NER) pathway,33 a DNA repair pathway that is disrupted in BRCA1-associated breast cancers. Deletions overlapping GTF2H2 are associated with decreased risk of breast cancer, suggesting that disruption of NER may be protective against the biological consequences of a BRCA1 pathogenic variant. The potential biological effect of the remaining deletions is unclear.
Genetic associations identified by this study included rare (<1% MAF) and polymorphic (>1% MAF) deletions that occurred at relatively low frequency (<10%) within the study cohort. Notably, no deletion polymorphism was observed overlapping the APOBEC3 locus, which has previously been associated with risk of both breast and ovarian cancer.11, 12, 13 This might be expected as the Illumina 610k array contains only two probes across the CNV region located between the fifth exon of APOBEC3A and the eighth exon of APOBEC3B so the variant is unlikely to be detected.12 Although this study identifies CNVs in BRCA1 pathogenic variant carriers, the low frequency of CNVs (all <10% in this study) and sample size limits the power to detect association in this study, in particular no associations reported here are significant after controlling for a false discovery rate of 0.05.34 Replication of CNVs identified by this study using larger data sets will be required to verify these associations. Moreover, larger cohort sizes will facilitate more detailed analyses to be performed, such as competing risks analyses to evaluate the associations with breast and ovarian cancer risks simultaneously. Importantly, genotyping data currently being derived by the large Oncoarray Network containing DNA samples from ~20 000 BRCA1 pathogenic variant carriers (http://epi.grants.cancer.gov/oncoarray/) will enable additional genome-wide CNV analysis and further assessment of candidate gene regions identified by this study.
Change history
22 August 2018
This Article was originally published under a CC BY-NC-SA 4.0 license, but has now been made available under a CC BY 4.0 license. The PDF and HTML versions of the Article have been modified accordingly.
References
Mavaddat N, Peock S, Frost D et al: Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 2013; 105: 812–822.
Milne RL, Antoniou AC : Genetic modifiers of cancer risk for BRCA1 and BRCA2 mutation carriers. Ann Oncol 2011; 22: i11–i17.
Couch FJ, Wang X, McGuffog L et al: Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet 2013; 9: e1003212.
Gaudet MM, Kuchenbaecker KB, Vijai J et al: Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS Genet 2013; 9: e1003173.
Bojesen SE, Pooley KA, Johnatty SE et al: Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet 2013; 45: 371–384, 384e1–2.
Kuchenbaecker KB, Neuhausen SL, Robson M et al: Associations of common breast cancer susceptibility alleles with risk of breast cancer subtypes in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res 2014; 16: 3416.
Zarrei M, MacDonald JR, Merico D, Scherer SW : A copy number variation map of the human genome. Nat Rev Genet 2015; 16: 172–183.
Stranger BE, Forrest MS, Dunning M et al: Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007; 315: 848–853.
Fanciulli M, Petretto E, Aitman TJ : Gene copy number variation and common human disease. Clin Genet 2010; 77: 201–213.
Krepischi AC, Pearson PL, Rosenberg C : Germline copy number variations and cancer predisposition. Future Oncol 2012; 8: 441–450.
Qi G, Xiong H, Zhou C : APOBEC3 deletion polymorphism is associated with epithelial ovarian cancer risk among Chinese women. Tumour Biol 2014; 35: 5723–5726.
Long J, Delahanty RJ, Li G et al: A common deletion in the APOBEC3 genes and breast cancer risk. J Natl Cancer Inst 2013; 105: 573–579.
Xuan D, Li G, Cai Q et al: APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry. Carcinogenesis 2013; 34: 2240–2243.
Antoniou AC, Wang X, Fredericksen ZS et al: A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet 2010; 42: 885–892.
Wang K, Li M, Hadley D et al: PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007; 17: 1665–1674.
Colella S, Yau C, Taylor JM et al: QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res 2007; 35: 2013–2025.
Frank B, Bermejo JL, Hemminki K et al: Copy number variant in the candidate tumor suppressor gene MTUS1 and familial breast cancer risk. Carcinogenesis 2007; 28: 1442–1445.
Walsh T, Casadei S, Coats KH et al: Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 2006; 295: 1379–1388.
Diehn M, Sherlock G, Binkley G et al: SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res 2003; 31: 219–223.
Giardine B, Riemer C, Hardison RC et al: Galaxy: a platform for interactive large-scale genome analysis. Genome Res 2005; 15: 1451–1455.
Blankenberg D, Von Kuster G, Coraor N et al: Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol 2010; Chapter 19: Unit 19 10 11–Unit 19 10 21.
Goecks J, Nekrutenko A, Taylor J : Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010; 11: R86.
Barnes DR, Lee A, Easton DF, Antoniou AC : Evaluation of association methods for analysing modifiers of disease risk in carriers of high-risk mutations. Genet Epidemiol 2012; 36: 274–291.
Dialsingh I, Austin SR, Altman NS : Estimating the proportion of true null hypotheses when the statistics are discrete. Bioinformatics 2015; 31: 2303–2309.
Walker LC, Wiggins GAR, Pearson JF : The role of constitutional copy number variants in breast cancer. Microarrays 2015; 4: 17.
ENCODE Project Consortium: An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Roadmap Epigenomics C, Kundaje A, Meuleman W et al: Integrative analysis of 111 reference human epigenomes. Nature 2015; 518: 317–330.
Hnisz D, Abraham BJ, Lee TI et al: Super-enhancers in the control of cell identity and disease. Cell 2013; 155: 934–947.
Nakano M, Fukushima Y, Yokota S et al: CYP2A7 pseudogene transcript affects CYP2A6 expression in human liver by acting as a decoy for miR-126. Drug Metabo Dispos 2015; 43: 703–712.
Xu C, Goodz S, Sellers EM, Tyndale RF : CYP2A6 genetic variation and potential consequences. Adv Drug Deliv Rev 2002; 54: 1245–1256.
Liu YL, Xu Y, Li F, Chen H, Guo SL : CYP2A6 deletion polymorphism is associated with decreased susceptibility of lung cancer in Asian smokers: a meta-analysis. Tumour Biol 2013; 34: 2651–2657.
Cancer Genome Atlas Research Network: Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–615.
Mjelle R, Hegre SA, Aas PA et al: Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair 2015; 30: 53–67.
Benjamini Y, Yekutieli D : The control of the false discovery rate in multiple testing under dependency. Ann Stat 2001; 29: 1165–1188.
Acknowledgements
LCW is funded by an HRC Sir Charles Hercus Health Research Fellowship. ABS is funded by an NHMRC Senior Research Fellowship. TO’M is funded by an NHMRC CJ Martin Early Career Fellowship.
BCFR: This work was supported by grant UM1 CA164920 from the USA National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products or organizations imply endorsement by the USA Government or the BCFR.
CIMBA: We wish to thank the patients and families, physicians, genetic counselors and research nurses involved in the CIMBA studies. The CIMBA data management and data analysis were supported by CRUK grants C12292/A11174 and C1287/A10118.
CNIO: We thank Alicia Barroso, Rosario Alonso and Guillermo Pita for their assistance. This work was partially supported by Spanish Association against Cancer (AECC08), RTICC 06/0020/1060, FISPI08/1120, Mutua Madrileña Foundation (FMMA) and SAF2010-20493.
COH-CCGCRN: Patients were recruited for study from the City of Hope Clinical Cancer Genomics Community Research Network, supported in part by Award Number RC4CA153828 (PI: J Weitzel) from the National Cancer Institute and the Office of the Director, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONSIT TEAM–INT: Monica Barile and Irene Feroce of the Istituto Europeo di Oncologia, Milan, Italy; Gabriele Capone of the University of Florence, Florence, Italy; Alessandra Viel and Riccardo Dolcetti of the CRO Aviano National Cancer Institute, Aviano (PN), Italy. Associazione Italiana Ricerca sul Cancro (AIRC) to P Radice (IG 2014 Id.15547). Funds from Italian citizens who allocated the 5 × 1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects ‘5 × 1000’).
EMBRACE: EMBRACE is supported by Cancer Research UK Grants C1287/A10118 and C1287/A11990. The Investigators at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust are supported by an NIHR grant to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust.
FCCC: We thank Ms JoEllen Weaver and Dr Betsy Bove for their technical support. The authors acknowledge support from The University of Kansas Cancer Center (P30 CA168524) and the Kansas Bioscience Authority Eminent Scholar Program. AKG was funded by 5U01CA113916, R01CA140323 and by the Chancellors Distinguished Chair in Biomedical Sciences Professorship.
GC-HBOC: The German Consortium of Hereditary Breast and Ovarian Cancer (GC-HBOC) is supported by the German Cancer Aid (grant no 110837, Rita K Schmutzler).
GEMO: Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO) study: National Cancer Genetics Network «UNICANCER Genetic Group», France. We wish to pay a tribute to Olga M Sinilnikova, who with Dominique Stoppa-Lyonnet initiated and coordinated GEMO until she sadly passed away on 30 June 2014, and to thank all the GEMO collaborating groups for their contribution to this study. GEMO Collaborating Centers are: Coordinating Centers, Unité Mixte de Génétique Constitutionnelle des Cancers Fréquents, Hospices Civils de Lyon - Centre Léon Bérard and Equipe «Génétique du cancer du sein»; Centre de Recherche en Cancérologie de Lyon: Olga Sinilnikova, Sylvie Mazoyer, Francesca Damiola, Laure Barjhoux, Carole Verny-Pierre, Mélanie Léone, Nadia Boutry-Kryza, Alain Calender and Sophie Giraud; and Service de Génétique Oncologique, Institut Curie, Paris: Dominique Stoppa-Lyonnet, Marion Gauthier-Villars, Bruno Buecher, Claude Houdayer, Etienne Rouleau, Lisa Golmard, Agnès Collet, Virginie Moncoutier, Muriel Belotti, Antoine de Pauw, Camille Elan, Catherine Nogues, Emmanuelle Fourme and Anne-Marie Birot. Institut Gustave Roussy, Villejuif: Brigitte Bressac-de-Paillerets, Olivier Caron and Marine Guillaud-Bataille. Centre Jean Perrin, Clermont–Ferrand: Yves-Jean Bignon and Nancy Uhrhammer. Centre Léon Bérard, Lyon: Christine Lasset, Valérie Bonadona and Sandrine Handallou. Centre François Baclesse, Caen: Agnès Hardouin, Pascaline Berthet, Dominique Vaur and Laurent Castera. Institut Paoli Calmettes, Marseille: Hagay Sobol, Violaine Bourdon, Tetsuro Noguchi, Audrey Remenieras and François Eisinger. CHU Arnaud-de-Villeneuve, Montpellier: Isabelle Coupier and Pascal Pujol. Centre Oscar Lambret, Lille: Jean-Philippe Peyrat, Joëlle Fournier, Françoise Révillion, Philippe Vennin and Claude Adenis. Centre Paul Strauss, Strasbourg: Danièle Muller and Jean-Pierre Fricker. Institut Bergonié, Bordeaux: Emmanuelle Barouk-Simonet, Françoise Bonnet, Virginie Bubien, Nicolas Sevenet and Michel Longy. Institut Claudius Regaud, Toulouse: Christine Toulas, Rosine Guimbaud, Laurence Gladieff and Viviane Feillel. CHU Grenoble: Dominique Leroux, Hélène Dreyfus, Christine Rebischung and Magalie Peysselon. CHU Dijon: Fanny Coron, Laurence Faivre. CHU St-Etienne: Fabienne Prieur, Marine Lebrun and Caroline Kientz. Hôtel Dieu Centre Hospitalier, Chambéry: Sandra Fert Ferrer. Centre Antoine Lacassagne, Nice: Marc Frénay. CHU Limoges: Laurence Vénat-Bouvet. CHU Nantes: Capucine Delnatte. CHU Bretonneau, Tours: Isabelle Mortemousque. Groupe Hospitalier Pitié-Salpétrière, Paris: Florence Coulet, Chrystelle Colas, Florent Soubrier and Mathilde Warcoin. CHU Vandoeuvre-les-Nancy: Johanna Sokolowska and Myriam Bronner. CHU Besançon: Marie-Agnès Collonge-Rame and Alexandre Damette. Creighton University, Omaha, USA: Henry T Lynch and Carrie L Snyder. The study was supported by the Ligue Nationale Contre le Cancer; the Association ‘Le cancer du sein, parlons-en!’ Award; the Canadian Institutes of Health Research for the ‘CIHR Team in Familial Risks of Breast Cancer’ program and the French National Institute of Cancer (INCa).
GEORGETOWN: Support was provided by the Nontherapeutic Subject Registry Shared Resource at Georgetown University (NIH/NCI grant P30-CA051008), the Fisher Center for Hereditary Cancer and Clinical Genomics Research and Swing Fore the Cure.
HCSC: We acknowledge Alicia Tosar and Paula Diaque for their technical assistance. HCSC was supported by a grant RD12/0036/0006 and 15/00059 from ISCIII (Spain), partially supported by European Regional Development FEDER funds.
HEBON: The Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) consists of the following Collaborating Centers: Coordinating center: Netherlands Cancer Institute, Amsterdam, Netherlands: MA Rookus, FBL Hogervorst, FE van Leeuwen, S Verhoef, MK Schmidt, NS Russell, JL de Lange and R Wijnands; Erasmus Medical Center, Rotterdam, Netherlands: JM Collée, AMW van den Ouweland, MJ Hooning, C Seynaeve, CHM van Deurzen and IM Obdeijn; Leiden University Medical Center, Netherlands: CJ van Asperen, JT Wijnen, RAEM Tollenaar, P Devilee and TCTEF van Cronenburg; Radboud University Nijmegen Medical Center, Netherlands: CM Kets and AR Mensenkamp; University Medical Center Utrecht, Netherlands: MGEM Ausems, RB van der Luijt and CC van der Pol; Amsterdam Medical Center, Netherlands: CM Aalfs and TAM van Os; VU University Medical Center, Amsterdam, Netherlands: JJP Gille, Q Waisfisz and HEJ Meijers-Heijboer; University Hospital Maastricht, Netherlands: EB Gómez-Garcia and MJ Blok; University Medical Center Groningen, Netherlands: JC Oosterwijk, AH van der Hout, MJ Mourits and GH de Bock; The Netherlands Foundation for the detection of hereditary tumors, Leiden, Netherlands: HF Vasen; The Netherlands Comprehensive Cancer Organization (IKNL): S Siesling and J Verloop; The Dutch Pathology Registry (PALGA): LIH Overbeek. The HEBON study is supported by the Dutch Cancer Society grants NKI1998-1854, NKI2004-3088 and NKI2007-3756, the Netherlands Organization of Scientific Research grant NWO 91109024, the Pink Ribbon grants 110005 and 2014-187.WO76, the BBMRI grant NWO 184.021.007/CP46, and the Transcan grant JTC 2012 Cancer 12-054. HEBON thanks the registration teams of IKNL and PALGA for part of the data collection.
ICO: We wish to thank the ICO Hereditary Cancer Program team led by Dr Gabriel Capella. Contract grant sponsor: Asociación Española Contra el Cáncer and Spanish Health Research Fund; Carlos III Health Institute; Catalan Health Institute; and Autonomous Government of Catalonia. Contract grant numbers: ISCIIIRETIC RD06/0020/1051, RD12/0036/008, PI10/01422, PI10/00748, PI13/00285, PIE13/00022, 2009SGR290 and 2014SGR364.
IHCC: The IHCC was supported by Grant PBZ_KBN_122/P05/2004.
IOVHBOCS: IOVHBOCS is supported by Ministero della Salute and ‘5 × 1000’ Istituto Oncologico Veneto grant.
KCONFAB: We wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab. kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia.
MAYO: MAYO is supported by NIH grants CA116167, CA192393 and CA176785, an NCI Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation and a generous gift from the David F and Margaret T Grohne Family Foundation.
MSKCC: We wish to thank Anne Lincoln and Lauren Jacobs. MSKCC is supported by grants from the Breast Cancer Research Foundation, the Robert and Kate Niehaus Clinical Cancer Genetics Initiative and the Andrew Sabin Research Fund.
SWE-BRCA: Swedish scientists participating as SWE-BRCA collaborators are from: Lund University and University Hospital: Åke Borg, Håkan Olsson, Helena Jernström, Karin Henriksson, Katja Harbst, Maria Soller and Ulf Kristoffersson; Gothenburg Sahlgrenska University Hospital: Anna Öfverholm, Margareta Nordling, Per Karlsson and Zakaria Einbeigi; Stockholm and Karolinska University Hospital: Anna von Wachenfeldt, Annelie Liljegren, Annika Lindblom, Brita Arver, Gisela Barbany Bustinza and Johanna Rantala; Umeå University Hospital: Beatrice Melin, Christina Edwinsdotter Ardnor and Monica Emanuelsson; Uppsala University: Hans Ehrencrona, Maritta Hellström Pigg and Richard Rosenquist; and Linköping University Hospital: Marie Stenmark-Askmalm and Sigrun Liedgren. SWE-BRCA collaborators are supported by the Swedish Cancer Society.
UCSF: We would like to thank Dr Robert Nussbaum and the following genetic counselors for participant recruitment: Beth Crawford, Kate Loranger, Julie Mak, Nicola Stewart, Robin Lee, Amie Blanco and Peggy Conrad. And thanks to Ms Salina Chan for her data management. Funding was provided by the UCSF Cancer Risk Program and Helen Diller Family Comprehensive Cancer Center.
UPENN: National Institutes of Health (NIH; R01-CA102776 and R01-CA083855; Breast Cancer Research Foundation; Susan G. Komen Foundation for the cure, Basser Research Center for BRCA.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Walker, L., Marquart, L., Pearson, J. et al. Evaluation of copy-number variants as modifiers of breast and ovarian cancer risk for BRCA1 pathogenic variant carriers. Eur J Hum Genet 25, 432–438 (2017). https://doi.org/10.1038/ejhg.2016.203
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.203
This article is cited by
-
Associating CYP2A6 structural variants with ovarian and lung cancer risk in the UK Biobank: replication and extension
European Journal of Human Genetics (2024)
-
Characterization of large-scale genomic differences in the first complete human genome
Genome Biology (2023)
-
Genotyping, characterization, and imputation of known and novel CYP2A6 structural variants using SNP array data
Journal of Human Genetics (2023)
-
Copy number variants as modifiers of breast cancer risk for BRCA1/BRCA2 pathogenic variant carriers
Communications Biology (2022)
-
Screening of BRCA1/2 genes mutations and copy number variations in patients with high risk for hereditary breast and ovarian cancer syndrome (HBOC)
BMC Cancer (2020)
