
Abstract
The medial prefrontal cortex (mPFC) is known to regulate executive decisions and the expression of emotional memories. More specifically, the prelimbic cortex (PL) of the mPFC is implicated in driving emotional responses via downstream targets including the nucleus accumbens and amygdala, but mechanisms are yet to be fully understood. Therefore, we investigated whether prelimbic cortical brain-derived neurotrophic factor (BDNF) signaling through the high-affinity tyrosine kinase receptor B (TrkB) receptor may serve as a molecular mechanism underlying emotional memory encoding. Here, we utilized viral-mediated inducible bdnf deletion within the PL, as well as TrkBF616A mutant mice, wherein TrkB receptor point mutation results in its being highly sensitive to inhibition by small PP1-derivative molecules, serving as a specific TrkB inhibitor. The site-specific TrkB antagonism and viral-mediated bdnf deletion within the PL resulted in deficits in both cocaine-dependent associative learning and fear expression. Deficiencies were rescued by the novel TrkB agonist 7,8-dihydroxyflavone, indicating that PL BDNF expression and downstream signaling through the TrkB receptor are required for memory formation in both appetitive and aversive domains.
Similar content being viewed by others


Introduction
The formation of aversively and appetitively valenced memories is critical for normal behavior. However, such memories can become detrimental to health if they evolve into uncontrolled fear, anxiety or reward seeking. The medial prefrontal cortex (mPFC) is highly implicated in both aversive and appetitive learning.1 Of particular interest is the prelimbic cortical subregion (PL), which sends robust projections to the basolateral nucleus of the amygdala and nucleus accumbens,2 providing clear connectivity to fear and reward pathways. A firmer grasp on the neurobiology of the PL is critical for understanding the mechanisms of normal learning and the development of fear and anxiety disorders such as post-traumatic stress disorder, as well as drug addiction and co-morbidities.
Activation of the PL is required for the expression of previously learned fears.1 Additionally, PL neurons undergo plasticity following fear-conditioning, during the presumptive consolidation period.3, 4, 5 Conversely, PL inactivation reduces freezing behavior in fear-conditioned rats,6 suggesting that PL is necessary for the expression of previously learned fear. The molecular mechanisms that regulate fear expression, consolidation and extinction are, however, still unresolved.
One promising mechanism is brain-derived neurotrophic factor (BDNF), and its high-affinity receptor tyrosine kinase receptor B (TrkB). Considerable evidence indicates that BDNF has an important role in regulating appetitive and aversive learning,7, 8 particularly in the prefrontal cortex.9, 10, 11 However, more remains to be determined about the role of BDNF and TrkB signaling in the PL, specifically with regard to emotional memory. Here, we elucidate a molecular mechanism involving BDNF signaling through TrkB in the PL,which regulates the expression of both appetitive and aversive emotional memories, providing compelling evidence that BDNF is a ‘master regulator’ of emotional memory in this prefrontal cortical region.
Materials and methods
Animals
Conditional mutant mice
Lentivirus experiments used homozygous bdnf-floxed mice (originally obtained from Jackson Labs, Bdnftm3Jae/J (B6/129S4/BALB/C background) and bred within our animal facility). These mice express loxP sites both upstream and downstream of exon 5 of the BDNF gene.12 TrkB F616A mutant mice (129J/C57Bl/6 hybrid background): The TrkB KI mice have a single-point mutation within the ATP-binding pocket of the kinase subdomain V of TrkB receptor, resulting in sensitivity to inhibition by small PP1-derivative molecules including 1-NM-PP1.13
All experiments were performed with adult (2–4 months old) male mice, which were group-housed in a temperature-controlled vivarium, with ad libitum access to food and water. They were maintained on a 12 h light/dark cycle, with all behavioral procedures being performed during the light cycle. All procedures used were approved by the Institutional Animal Care and Use Committee of Emory University, and in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals.
Pharmacological drugs
TrkBF616A mutant mice were unilaterally implanted with guide cannulae targeting the PL (internal cannulae tip coordinates from Bregma: AP +2.0, ML±0.4, DV −2.0) (n=12 per group) using stereotaxic surgery under ketamine (75 mg/kg)/dormitor (1 mg/kg) anesthesia. TrkB KI mice were bilaterally infused with 0.1 nmol 1-NM-PP1 (TrkB inhibitor) (0.2 mM in 4% dimethylsulfoxide, 2% Tween-20) or vehicle over 1 min immediately following fear-conditioning. 7,8-dihydroxflavone (7,8-DHF) was dosed systemically intraperitoneally (i.p.) at a 5 mg/kg dose in 17% dimethylsulfoxide in phosphate-buffered saline as previously described.10, 14, 15 The injection volume was 1 ml/100 g.
Cre-recombinase lentivirus infection
Cre-recombinase expressing vector (LV-Cre) or a greenfluorescent protein (GFP)-expressing control vector (LV-GFP), delta8.9 and VSV-g were co-transfected into HEK293T producer cells to produce replication-incompetent but highly infective virus. The packaged virus was concentrated through a number of ultracentrifugation steps as described previously16, 17, 18 Viruses were titred to reach at least 1 × 109 infectious particles per ml. Green fluorescent protein-expressing control vector (n=12) or cre-recombinase expressing vector (n=12) virus was bilaterally injected using a 26-gauge Hamilton syringe (precoated with 10 mg/ml Bovine Serum Albumin) on a microinjection pump (0.25 μl/15 min) into the PL (coordinates from Bregma: AP +2.0, ML±0.3, DV −2.0) of homozygous floxed BDNF mice using stereotaxic surgery under ketamine (75 mg/kg)/dormitor (1 mg/kg) anesthesia. Lentiviral infections were confirmed by GFP fluorescence, and in situ hybridization for Cre-recombinase and BDNF mRNA (see below). Mice in which the spread of infection was localized bilaterally, in the PL, and with limited to no spread into adjacent cortical subregions were included in this study (n=7 per group).
Cue-dependent fear conditioning
All mice were preexposed three times to the startle chambers (San Diego Instruments, San Diego, CA, USA) for 10 min of habituation, 3 days before training (14 days following surgeries for lentivirus-infected mice). During cued fear training, mice received five paired conditioned stimulus tone (30 s, 6 kHz, 90 db) or (30 s, 12 kHz, 80 db) co-terminating with unconditioned stimulus shock (500 ms, 1.0 mA) trials with a 5 min inter-trial interval in the startle boxes. Startle response to the shocks, and percent time spent freezing to the tones was measured by SR-LAB software (San Diego Instruments).
Cued fear expression test
The expression of fear memory was tested 24 h after fear-conditioning in a novel context (altering flooring with mats/sand paper, lights on vs lights off, and/or square vs circular walls) (modular test chambers; MedAssociates, St Albans, VT, USA). The mice were exposed to 15 conditioned stimulus tones with a 1.5 min inter-trial interval, and freezing during the tone presentations was measured with FreezeView software (Coulbourn Instruments, Allentown, PA, USA).
Fear extinction training and retention tests
Extinction training occurred 24 h after conditioning, and extinction retention test occurred 24 h after extinction training similarly in the modular test chambers except that the mice were exposed to 30 conditioned stimulus tones with a 30-s inter-trial interval for each test session.
Cocaine-conditioned place preference
Conditioned place preference (CPP) apparatuses, (MedAssociates) each consisted of two conditioning chambers (black with rod floor and white with mesh floor) connected by a central chamber with vertical sliding manual doors. The chamber-adjustable light levels were optimized for balanced preference between black and white conditioning chambers. Fifteen photobeams recorded the time spent in each compartment. The mice received a pretraining test for individual place preference by placing the mouse in the neutral compartment and allowing 20 min of free access to all three chambers. The day after pretesting, CPP training took place over 3 consecutive days, during which each day animal received one cocaine-paired session (10 mg per kg of cocaine-hydrochloride i.p. (Sigma-Aldrich, St Louis, MO, USA) dissolved in saline) in the white chamber (least preferred chamber during pretest), and one saline vehicle-pairing session in the black chamber (distributed and alternating in the 0900–1100 h and 1400–1600 h sessions) for 30 min. The day after the completion of CPP training, all the mice received posttraining test (identical to pretraining test), in which animals were allowed to freely explore all three chambers for 20 min. Cocaine-associated place preference data were calculated as percent time spent on cocaine-paired chamber over the total time spent in the two conditioning chambers.
Open field, locomotor sensitivity test
The open field was an open box (27.9 cm × 27.9 cm) made of Plexiglass. The mice were placed in the apparatus to explore for 10 min and then returned to home cages. Activity data were obtained and analyzed using the Activity Software (Med Associates.).
Locomotor sensitivity to psychostimulant administration was also tested using similar methods. Mice were placed in customized Med-Associates locomotor chambers equipped with 16 photobeams, which were broken as the animal moved throughout the cage. Mice were initially habituated to the chamber for 1 h. The following day, mice were again allowed to explore the chamber for 1 h, after which methylphenidate (5 mg/kg, i.p., Sigma-Aldrich) was injected. The dose was selected to match the dose of cocaine for efficacy at the dopamine transporter. Ambulation and repeated interruption of the same photobeam, suggestive of stereotypy, were monitored for an additional hour; the photobeams broken after injection were normalized to a pretest baseline to account for individual differences between mice.
Tissue preparation
Mice were killed by decapitation, and the brains were collected and flash frozen on dry ice and stored at −80 °C until sectioned on a cryostat into six parallel series of brain sections on microscope slides (16 μM/section). Brain sections were stored at −80 °C until used for in situ hybridization.
In situ hybridization
Region-specific Cre gene expression and bdnf deletion were confirmed with in situ hybridization. In brief, pretreatment of tissue slides included fixation in 4% paraformaldehyde, proteinase K digestion and acetylation as we have previously done.19 The slides were hybridized with 35S-UTP-labeled cRNA riboprobes, prepared from linearized constructs for antisense sequences of BDNF (exon 5), using T7 RNA polymerase and Cre-recombinase with SP6 RNA polymerase at 50 °C overnight. Posthybridization included RNase A treatment, stringent SSC washes and dehydration. Slides were exposed to on Biomax MR autoradiography film (Eastman Kodak , Rochester, NY, USA, for 1 day (Cre), for 14 days (BDNF). Developed film images were scanned into high-resolution image files. Site and spread of the virus infection were localized by Cre mRNA or by GFP. Mice with infection sites outside of the prelimbic cortex were removed from all statistical analyses.
Statistical analyses
Fear acquisition, expression and extinction data as well as cocaine preference data were analyzed by two-way analysis of variance with repeated measures. All other behavioral tests were analyzed by one-way analysis of variance. Statistically significant main effects or interactions were followed by post-hoc least squares difference tests for multiple comparisons. Data are presented as mean±s.e.m., and statistical significance was set at P<0.05. Tests for homogeneity of variance were performed and when necessary, square root transformations were used. Detection of outliers was performed, and when necessary, removed from analyses, and data were reanalyzed after outlier removal and/or transformation.
Results
We utilized conditional mutant homozygous BDNF-floxed mice12 and lentivirus-expressing Cre-recombinase to knockdown bdnf bilaterally in the PL (Figures 1a–c). Two weeks following surgery, the mice were fear conditioned to five trials of conditioned stimulus tone (30 s, 6 kHz, 90 db) co-terminating with unconditioned stimulus shock (500 ms, 1.0 mA) (Figure 2a). The following day, mice with site-specific deletions had robustly attenuated expression of learned fear to the cue (F1,48=3.79; P<0.05) (Figure 2b). These mice were then fear extinction trained until there were no differences in freezing to the cue. They were subsequently retrained with a novel cue (12 kHz tone) (with notably no differences in freezing during retraining (data not shown)), immediately followed by a systemic infusion of a potent TrkB agonist, 7,8-DHF (5 mg/kg).10, 15, 20 One day after retraining to the novel cue followed by TrkB agonist administered during the fear consolidation period, PL bdnf knockdown mice now expressed fear similarly to the control mice, P>0.05 (Figure 2c), indicating that driving TrkB signaling is sufficient to rescue fear learning deficits in bdnf knockdown mice.

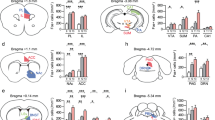
Representative images of site-specific inducible PL bdnf knockdown. (a) Cre-recombinase mRNA expression at the site of infection. (b) Selective deletion of bdnf mRNA in the adjacent hemisphere. Note that the infralimbic cortex is largely spared. (c) Representative spread of infection in these experiments, with gray indicating the largest spread and black the smallest.

Site-specific PL bdnf knockdown attenuates cued fear-conditioning: Rescue by a TrkB agonist. (a) PL bdnf knockdown mice have normal acquisition of fear. (b) PL bdnf knockdown mice have reduced sensitivity to the fear-conditioning, as indicated by less freezing following cued fear-conditioning. (c) Systemic TrkB agonist 7,8-DHF rescued fear-associated learning, as there were no differences in freezing between groups. *P<0.05. ‘LV-Cre’ refers to knockdown mice.
We next investigated whether bdnf knockdown has its behavioral effects via diminished signaling through PL TrkB. We used TrkBF616A mutant mice (TrkB KI), which have a single-point mutation, resulting in sensitive inhibition by small PP1-derivative molecules including 1-NM-PP1.13 TrkB KI mice were cannulated, received cued fear-conditioning training and immediately followed infusion with the TrkB inhibitor 1-NM-PP1 (0.1 nmol) or vehicle within PL (Figure 3a). Only TrkB KI mice that received 1-NM-PP1 had attenuated freezing to the cue (F1,13=14.03,P<0.05) (Figure 3b). After extinction training and retraining to a novel cue in the absence of the inhibitor, the same mice expressed fear equally well, P>0.05 (Figure 3c).

Site-specific PL TrkB inhibitor attenuates cued fear-conditioning. (a) Representative internal cannula placements targeting the PL. (b) TrkB inhibitor (1-NM-PP1 in F616 mice) also attenuated the consolidation of cued fear. (c) Retraining to a new cue in the absence of the inhibitor revealed no differences between groups. *P<0.05. ‘LV-Cre’ refers to knockdown mice.
To address the role of PL BDNF in appetitive learning, we trained PL-specific bdnf knockdown mice in a cocaine-CPP paradigm. Following training,the mice with PL bdnf deletions had attenuated preference for the cocaine-associated chamber (t(22)=23.90;P<0.05) (Figure 4a). Mice were retrained using novel contexts, with 7,8-DHF administered immediately posttraining. As in aversive domains, TrkB activation rescued cocaine-associated place preference (P>0.05) (Figure 4b).

PL bdnf knockdown blunt psychostimulant sensitivity. (a) Cocaine-CPP is diminished in knockdown mice. (b) Administration of a TrkB agonist rescues cocaine-dependent associative learning. (c) Locomotor sensitivity to methylphenidate was also blunted in bdnf knockdown mice, such that both ambulatory and stereotypy-like counts were reduced. *P<0.05. ‘LV-Cre’ refers to knockdown mice.
In the relatively confined space of the CPP chambers, we saw no differences in locomotor activity at the baseline or in response to cocaine (P>0.05, not shown). We also, however, tested locomotor sensitivity to methyphenidate at a dose matched to cocaine for efficacy at the dopamine transporter (5 mg/kg). In large locomotor chambers that allowed for considerable movement, bdnf knockdown decreased both methyphenidate-elicited ambulation and stereotypy-like counts (F1,28=5.6, P<0.05), providing evidence that bdnf knockdown interferes with dopamine- and norepinephrine-dependent mechanisms of psychostimulant action. As in our CPP chambers, however, there were no group differences in baseline locomotor counts (P>0.05, not shown).
Discussion
The PL bdnf gene deletion here resulted in attenuated fear responses following fear-conditioning, is consistent with the previous work, suggesting PL BDNF is required for consolidation of learned fear.10 In these bdnf knockdown mice, there were also no effects of bdnf silencing on the acquisition of fear during fear-conditioning, and there were no differences in the extinction of fear (data not shown), suggesting that PL BDNF is critical for consolidation of learned fear, but not extinction of fear. Additionally, this fear deficit was rescued by systemic administration of the TrkB agonist 7,8-DHF, indicating that TrkB activation during the consolidation period (following conditioning) is sufficient to rescue the knockdown-induced deficiencies. Our fear responses (% freezing) were not high enough to suggest a ceiling effect, and the TrkB agonist did not elevate fear expression in controls, suggesting normative fear expression cannot be further enhanced with the TrkB agonist. It is notable though, that we have previously reported that the same TrkB agonist enhances fear consolidation in wild-type mice,15 but this may be due to strain differences between C57 wild-type mice and bdnf-floxed mice, which are bred on a mixed 129S4/BALB/C background.
Toward determining whether BDNF acted specifically on TrkB receptors in the PL, we found that TrkB inhibition in the PL immediately following conditioning (that is, during the fear consolidation period) resulted in similar deficits in learned fear response. These findings indicate that TrkB signaling in PL is required for fear memory consolidation, and that PL circuitry retains the potential for new plasticity following transient TrkB inhibition. These findings do not preclude the possibility, however, that anterograde or retrograde PL BDNF transport also has a role in the expression of emotionally-valenced memories.21, 22 Notably, Peters et al.23 demonstrated in a parallel fashion that BDNF infusions into the infralimbic cortex of the medial prefrontal cortex could extinguish fear, and that hippocampal BDNF is critical for supplying and regulating the prefrontal cortical TrkB activity.
We also found that PL bdnf deletions were effective in blunting cocaine-CPP following CPP training, providing evidence that PL BDNF is critical for the consolidation of stimulus–outcome associative relationships in both aversive and appetitive domains. These bdnf knockdown mice had no differences in the extinction of cocaine-associated place preference in the absence of cocaine (data not shown), similar to the fear extinction, suggesting that PL BDNF is not critical for the extinction of Pavlovian drug- or fear-related associations. Interesting, PL BDNF is, however, essential for the extinction of operant responding for food outcomes,11 suggesting that the influence of PL BDNF differs according to associative—that is, Pavlovian vs operant—contexts.
Here, the TrkB agonist 7,8-DHF was sufficient to rescue the deficit in appetitive learning in PL bdnf knockdown mice, indicating the specificity of BDNF and TrkB in modulating cocaine-dependent associative learning. In contrast, BDNF overexpression in the dorsally situated anterior cingulate cortex suppresses cocaine seeking in models of relapse.9 The same is true of infusions into the NAc24 and VTA.25 Together, these data indicate that BDNF TrkB signaling has critical roles in emotional learning, but that these are highly region- or circuit-specific.
It has been previously reported that PL bdnf knockdown does not affect baseline locomotor activity,10 and we replicated this finding during the habituation of mice to our CPP testing chambers (data not shown). Therefore, we believe, PL bdnf knockdown behavioral findings are not confounded by differences in locomotion during freezing or cocaine-CPP tests, and that knockdown is therefore specifically affecting the consolidation of emotional memories.
As our findings using local inhibition of TrkB suggest, BDNF may act directly via TrkB receptors within the PL, but it is also likely that BDNF is projecting to terminals in downstream regions such as the amygdala, nucleus accumbens or ventral tegmental area.22, 26 BDNF effects via its TrkB receptor are likely anatomically selective, as BDNF infusions both dorsal and ventral to the PL have dissimilar consequences9, 23 likely due to the unique anatomy of PL projections.2
Overall, our data provide compelling evidence that PL BDNF is a master regulator of memory formation in multiple biological contexts. The PL is of critical importance, as it is positioned to integrate information regarding appetitive and aversive memories, and to subsequently modulate responses to emotional stimuli via targeting, for example, the nucleus accumbens and the basolateral amygdala, both of which are major downstream targets. A more complete perspective on neurotrophin actions within the medial prefrontal cortex will clarify the therapeutic approaches to affective disease states characterized by debilitating fear, anxiety and drug addiction co-morbidities.
References
Peters J, Kalivas PW, Quirk GJ . Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem 2009; 16: 279–288.
Vertes RP . Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 2004; 51: 32–58.
Laviolette SR, Lipski WJ, Grace AA . A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input. J Neurosci 2005; 25: 6066–6075.
Baeg EH, Kim YB, Jang J, Kim HT, Mook-Jung I, Jung MW . Fast spiking and regular spiking neural correlates of fear conditioning in the medial prefrontal cortex of the rat. Cereb Cortex 2001; 11: 441–451.
Gilmartin MR, McEchron MD . Single neurons in the medial prefrontal cortex of the rat exhibit tonic and phasic coding during trace fear conditioning. Behav Neurosci 2005; 119: 1496–1510.
Corcoran KA, Quirk GJ . Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci 2007; 27: 840–844.
Musumeci G, Minichiello L. BDNF-TrkB . signalling in fear learning: from genetics to neural networks. Rev Neurosci 2011; 22: 303–315.
McGinty JF, Whitfield TW, Berglind WJ . Brain-derived neurotrophic factor and cocaine addiction. Brain Res 2010; 1314: 183–193.
Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Miller SW et al. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci 2007; 26: 757–766.
Choi DC, Maguschak KA, Ye K, Jang SW, Myers KM, Ressler KJ . Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci USA. 2010; 107: 2675–2680.
Gourley SL, Howell JL, Rios M, DiLeone RJ, Taylor JR . Prelimbic cortex BDNF knock-down reduces instrumental responding in extinction. Learn Mem 2009; 16: 756–760.
Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 2001; 15: 1748–1757.
Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C et al. A chemical-genetic approach to studying neurotrophin signaling. Neuron 2005; 46: 13–21.
Andero R, Daviu N, Escorihuela RM, Nadal R, Armario A . 7,8-dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus 2012; 22: 399–408.
Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ . Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry 2011; 168: 163–172.
Rattiner LM, Davis M, French CT, Ressler KJ . Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci 2004; 24: 4796–4806.
Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ., Amygdala BDNF . signaling is required for consolidation but not encoding of extinction. Nat Neurosci 2006; 9: 870–872.
Heldt SA, Stanek L, Chhatwal JP, Ressler KJ . Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry 2007; 12: 656–670.
Rattiner LM, Davis M, Ressler KJ . Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem 2004; 11: 727–731.
Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA. 2010; 107: 2687–2692.
Sobreviela T, Pagcatipunan M, Kroin JS, Mufson EJ . Retrograde transport of brain-derived neurotrophic factor (BDNF) following infusion in neo- and limbic cortex in rat: relationship to BDNF mRNA expressing neurons. J Comp Neurol 1996; 375: 417–444.
Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997; 389: 856–860.
Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ . Induction of fear extinction with hippocampal-infralimbic BDNF. Science 2010; 328: 1288–1290.
Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW . Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci 2007; 10: 1029–1037.
Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y . A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci 2004; 24: 1604–1611.
Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S . Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci 1997; 17: 2295–2313.
Acknowledgements
This study was supported by the National Institutes of Health (F32MH085443, R01DA01962), the Center for Behavioral Neuroscience (NSF agreement IBN-987675), the Burroughs Wellcome Fund, the National Institutes of Health/NCRR base grant (P51RR000165) to Yerkes National Primate Research Center, and Children’s Health care of Atlanta.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare conflict of interests.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Choi, D., Gourley, S. & Ressler, K. Prelimbic BDNF and TrkB signaling regulates consolidation of both appetitive and aversive emotional learning. Transl Psychiatry 2, e205 (2012). https://doi.org/10.1038/tp.2012.128
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.128
Keywords
This article is cited by
-
Reinstatement of nicotine conditioned place preference in a transgenerational model of drug abuse vulnerability in psychosis: Impact of BDNF on the saliency of drug associations
Psychopharmacology (2023)
-
BDNF signaling during the lifetime of dendritic spines
Cell and Tissue Research (2020)
-
Estrogen-dependent association of HDAC4 with fear in female mice and women with PTSD
Molecular Psychiatry (2018)
-
Direct dorsal hippocampal–prelimbic cortex connections strengthen fear memories
Nature Neuroscience (2017)
-
Going and stopping: dichotomies in behavioral control by the prefrontal cortex
Nature Neuroscience (2016)
