
Abstract
Moyamoya disease (MMD) shows progressive cerebral angiopathy characterized by bilateral internal carotid artery stenosis and abnormal collateral vessels. Although ∼15% of MMD cases are familial, the MMD gene(s) remain unknown. A genome-wide association study of 785 720 single-nucleotide polymorphisms (SNPs) was performed, comparing 72 Japanese MMD patients with 45 Japanese controls and resulting in a strong association of chromosome 17q25-ter with MMD risk. This result was further confirmed by a locus-specific association study using 335 SNPs in the 17q25-ter region. A single haplotype consisting of seven SNPs at the RNF213 locus was tightly associated with MMD (P=5.3 × 10−10). RNF213 encodes a really interesting new gene finger protein with an AAA ATPase domain and is abundantly expressed in spleen and leukocytes. An RNA in situ hybridization analysis of mouse tissues indicated that mature lymphocytes express higher levels of Rnf213 mRNA than their immature counterparts. Mutational analysis of RNF213 revealed a founder mutation, p.R4859K, in 95% of MMD families, 73% of non-familial MMD cases and 1.4% of controls; this mutation greatly increases the risk of MMD (P=1.2 × 10−43, odds ratio=190.8, 95% confidence interval=71.7–507.9). Three additional missense mutations were identified in the p.R4859K-negative patients. These results indicate that RNF213 is the first identified susceptibility gene for MMD.
Similar content being viewed by others
Introduction
‘Moyamoya’ is a Japanese expression for something hazy, such as a puff of cigarette smoke drifting in the air. In individuals with Moyamoya disease (MMD), there is a progressive stenosis of the internal carotid arteries; a fine network of collateral vessels, which resembles a puff of smoke on a cerebral angiogram, develops at the base of the brain (Figure 1a).1, 2 This steno-occlusive change can cause transient ischemic attacks and/or cerebral infarction, and rupture of the collateral vessels can cause intracranial hemorrhage. Children under 10 years of age account for nearly 50% of all MMD cases.3

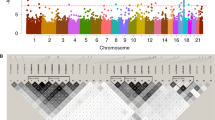
(a) Abnormal brain vessels in MMD. The dotted circle indicates the X-ray field of cerebral angiography (left panel). Normal structures of the right internal carotid artery (ICA), anterior cerebral artery (ACA) and middle cerebral artery (MCA) are illustrated (middle panel). The arrowheads indicate abnormal collateral vessels appearing like a puff of smoke in the angiogram of an individual with MMD (right panel). Note that ACA and MCA are barely visible, because of the occlusion of the terminal portion of the ICA. (b) Manhattan plot of the 785 720 SNPs used in the genome-wide association analysis of MMD patients. Note that the SNPs in the 17q25-ter region reach a significance of P<10−8.
The etiology of MMD remains unclear, although epidemiological studies suggest that bacterial or viral infection may be implicated in the development of the disease.4 Growing attention has been paid to the upregulation of arteriogenesis and angiogenesis associated with MMD because chronic ischemia in other disease conditions is not always associated with a massive development of collateral vessels.5, 6 Several angiogenic growth factors are thought to have functions in the development of MMD.7
Several lines of evidence support the importance of genetic factors in susceptibility to MMD.8 First, 10–15% of individuals with MMD have a family history of the disease.9 Second, the concordance rate of MMD in monozygotic twins is as high as 80%.10 Third, the prevalence of MMD is 10 times higher in East Asia, especially in Japan (6 per 100 000 population), than in Western countries.3 Familial MMD may be inherited in an autosomal dominant fashion with low penetrance or in a polygenic manner.11 Linkage studies of MMD families have revealed five candidate loci for an MMD gene: chromosomes 3p24–26,12 6q25,13 8q13–24,10 12p12–1310 and 17q25.14 However, no susceptibility gene for MMD has been identified to date.
We collected 20 familial cases of MMD to investigate linkage in the five putative MMD loci. However, a definitive result was not obtained for any of the loci. We then hypothesized that there might be a founder mutation among Japanese patients with MMD because the prevalence of MMD is unusually high in Japan.15 Genome-wide and locus-specific association studies were performed and successfully identified a single gene, RNF213, linked to MMD. We report here a strong association between MMD onset and a founder mutation in RNF213, as well as the expression profiles of RNF213, in various tissues.
Materials and methods
Affected individuals
Genomic DNA was extracted from blood and/or saliva samples obtained from members of the families with MMD (Supplementary Figure 1), MMD patients with no family history and control subjects. All of the subjects were Japanese. MMD was diagnosed on the basis of guidelines established by the Research Committee on Spontaneous Occlusion of the Circle of Willis of the Ministry of Health and Welfare of Japan. This study was approved by the Ethics Committee of Tohoku University School of Medicine. Total RNA samples were purified from leukocytes using an RNeasy mini kit (Qiagen, Hilden, Germany) and used as templates for cDNA synthesis with an Oligo (dT)20 primer and SuperScript II reverse transcriptase according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA).
Linkage analysis
For the linkage analysis, DNA samples were genotyped for 36 microsatellite markers within five previously reported MMD loci using the ABI 373A DNA Sequencer (Applied Biosystems, Foster City, CA, USA). Pedigrees and haplotypes were constructed with the Cyrillic version 2.1 software (Oxfordshire, UK). Multipoint analyses were conducted using the GENEHUNTER 2 software (http://www.broadinstitute.org/ftp/distribution/software/genehunter/). Statistical analysis was performed with SPSS version 14.0J (SPSS, Tokyo, Japan).
Genome-wide and locus-specific association studies
A genome-wide association study was performed using a group of 72 MMD patients, which consisted of 64 patients without a family history of MMD and 8 probands of MMD families. The Illumina Human Omni-Quad 1 chip (Illumina, San Diego, CA, USA) was used for genotyping, and single-nucleotide polymorphisms (SNPs) with a genotyping completion rate of 100% were used for further statistical analysis (785 720 out of 1 140 419 SNPs). Genotyping data from 45 healthy Japanese controls were obtained from the database at the International HapMap Project web site. The 785 720 SNPs were statistically analyzed using the PLINK software (http://pngu.mgh.harvard.edu/~purcell/plink/index.shtml). For a locus-specific association study, we used 63 DNA samples consisting of 58 non-familial MMD patients and 5 probands of MMD families. A total of 384 SNPs within chromosome 17q25-ter were genotyped (Supplementary Table 1), using the GoldenGate Assay and a custom SNP chip (Illumina). Genotyping data for 45 healthy Japanese were used as a control. Case–control single-marker analysis, haplotype frequency estimation and significance testing of differences in haplotype frequency were performed using the Haploview version 3.32 program (http://www.broad.mit.edu/mpg/haploview/).
Mutation detection
Mutational analyses of RNF213 and FLJ35220 were performed by PCR amplification of each coding exon and putative promoter regions, followed by direct sequencing. Genomic sequence data for the two genes were obtained from the National Center for Biotechnology Information web site (http://www.ncbi.nlm.nih.gov/) for design of exon-specific PCR primers. RNF213 cDNA fragments were amplified from leukocyte mRNA for sequencing analysis. Sequencing of the PCR products was performed with the ABI BigDye Terminator Cycle Sequencing Reaction Kit using the ABI 310 Genetic Analyzer. Identified base changes were screened in control subjects. Statistical difference of the carrier frequency of each base change was estimated by Fisher's exact test (the MMD group vs the control group).
Quantitative PCR
MTC Multiple Tissue cDNA Panels (Clontech Laboratory, Madison, WI, USA) were the source of cDNAs from human cell lines, adult and fetal tissues. Mononuclear cells and polymorphonuclear cells were isolated from the fresh peripheral blood of healthy human adults using Polymorphprep (Cosmo Bio, Carlsbad, CA, USA). T and B cells were isolated from the fresh peripheral blood of healthy human adults using the autoMACS separator (Militeny Biotec, Bergisch Gladbach, Germany). Total RNA was isolated from these cells with the RNeasy Mini Kit (Qiagen) following the manufacturer's instructions. We reverse transcribed 100 ng samples of total RNA into cDNAs using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCRs were performed in a final volume of 20 μl using the FastStart TaqMan Probe Master (Rox) (Roche, Madison, WI, USA), 5 μl of cDNA, 10 μM of RNF- or GAPDH-specific primers and 10 μM of probes (Universal ProbeLibrary Probe #80 for RNF213 and Roche Probe #60 for GAPDH). All reactions were performed in triplicate using the ABI 7500 Real-Time PCR system (Applied Biosystems). Cycling conditions were 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. Real-time PCR data were analyzed by the SDS version 1.2.1 software (Applied Biosystems). We evaluated the relative level of RNF213 mRNA by determining the CT value, the PCR cycle at which the reporter fluorescence exceeded the signal baseline. GAPDH mRNA was used as an internal reference for normalization of the quantitative expression values.
Multiplex PCR
MTC Multiple Tissue cDNA Panels (Clontech) were the source of human cell lines and cDNAs from human adult and fetal tissues. Multiplex PCRs were performed in a final volume of 20 μl using the Multiplex PCR Master Mix (Qiagen), 2 μl of cDNA, a 2 μM concentration of RNF213 and a 10 μM concentration of GAPDH-specific primers. The samples were separated on a 2% agarose gel stained with ethidium bromide. Cycling conditions were 15 min at 94°C, followed by 30 cycles of 30 s at 94 °C, 30 s at 57 °C and 30 s at 72 °C. For normalization of the expression levels, we used GAPDH as an internal reference for each sample.
In situ hybridization (ISH) analysis
Paraffin-embedded blocks and sections of mouse tissues for ISH were obtained from Genostaff (Tokyo, Japan). The mouse tissues were dissected, fixed with Tissue Fixative (Genostaff), embedded in paraffin by proprietary procedures (Genostaff) and sectioned at 6 μm. To generate anti-sense and sense RNA probes, a 521-bp DNA fragment corresponding to nucleotide positions 470–990 of mouse Rnf213 (BC038025) was subcloned into the pGEM-T Easy vector (Promega, Madison, WI, USA). Hybridization was performed with digoxigenin-labeled RNA probes at concentrations of 300 ng ml−1 in Probe Diluent-1 (Genostaff) at 60 °C for 16 h. Coloring reactions were performed with NBT/BCIP solution (Sigma-Aldrich, St Louis, MO, USA). The sections were counterstained with Kernechtrot stain solution (Mutoh, Tokyo, Japan), dehydrated and mounted with Malinol (Mutoh). For observation of Rnf213 expression in activated lymphocytes, 10-week-old Balb/c mice were intraperitoneally injected with 100 μg of keyhole limpet hemocyanin and incomplete adjuvant and sacrificed in 2 weeks. The spleen of the mice was removed for Hematoxylin–eosin staining and ISH analyses.
Results
Using 20 Japanese MMD families, we reevaluated the linkage mapped previously to five putative MMD loci. No locus with significant linkage, Lod score >3.0 or NPL score >4.0 was confirmed (Supplementary Figure 2). We conducted a genome-wide association study of 72 Japanese MMD cases. Single-marker allelic tests comparing the 72 MMD cases and 45 controls were performed for 785 720 SNPs using χ2 statistics. These tests identified a single locus with a strong association with MMD (P<10−8) on chromosome 17q25-ter (Figure 1b), which is in line with the latest mapping data of a MMD locus.16 The SNP markers with P<10−6 are listed in Table 1. To confirm this observation, we performed a locus-specific association study. A total of 384 SNP markers (Supplementary Table 1) were selected within the chromosome 17q25-ter region and genotyped in a set of 63 MMD cases and 45 controls. The SNP markers demonstrating a high association with MMD (P<10−6) were clustered in a 151-kb region from base position 75 851 399–76 003 020 (SNP No.116–136 in Supplementary Table 1); this entire region was within the RNF213 locus (Figure 2a). A single haplotype determined by seven SNPs (SNP Nos.130–136 in Supplementary Table 1) that resided in the 3′ region of RNF213 was strongly associated with MMD onset (P=5.3 × 10−10). Analysis of the linkage disequilibrium block indicated that this haplotype was not in complete linkage disequilibrium with any other haplotype in this region (Supplementary Figure 3). These results strongly suggest that a founder mutation may exist in the 3′ part of RNF213.

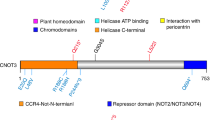
(a) Association analysis of 63 non-familial MMD cases and 45 control subjects. Statistical significance was evaluated by the χ2-test. SNP markers with a strong association with MMD (P<10−6) clustered in a 161-kb region (base position 75 851 399–76 012 838) indicated by two dotted lines (upper panel), which included the entire region of RNF213 (lower panel). Haplotype analysis revealed a strong association (P=5.3 × 10−10) between MMD and a single haplotype located within RNF213. (b) Sequencing chromatograms of the identified MMD mutations. The left panel shows the sequences of an unaffected individual and a carrier of a p.R4859K heterozygous mutation. The right panel indicates the sequencing chromatograms of the leukocyte cDNA obtained from an unaffected individual and an individual with MMD who carries the p.R4859K mutation. Note that both wild-type and mutant alleles were expressed in leukocytes. (c) The structure of the RNF213 protein. The RNF213 protein contains three characteristic structures, the AAA-superfamily ATPase motif, the RING motif and the HMG-CoA reductase degradation motif. The positions of four mutations identified in MMD patients are underlined, including one prevalent mutation (red) and three private mutations (black).
Mutational analysis of the entire coding and promoter regions of RNF213 and FLJ35220, a gene 3′ adjacent to RNF213, revealed that 19 of the 20 MMD families shared the same single base substitution, c.14576G>A, in exon 60 of RNF213 (Figure 2b and Table 2). This nucleotide change causes an amino-acid substitution from arginine4859 to lysine4859 (p.R4859K). The p.R4859K mutation was identified in 46 of 63 non-familial MMD cases (73%), including 45 heterozygotes and a single homozygote (Table 3). Both the wild-type and the p.R4859K mutant alleles were co-expressed in leukocytes in three patients heterozygous for the p.R4859K mutation (Figure 2b), excluding the possible instability of the mutant RNF213 mRNA. Additional missense mutations, p.M3940V, p.V4616M and p.V4814M, were detected in three non-familial MMD cases without the p.R4859K mutation (Figure 2c). These mutations were not found in 388 control subjects and were detected in only one patient, suggesting that they were private mutations (Table 2). No copy number variation or mutation was identified in the RNF213 locus of 12 MMD patients using comparative genome hybridization microarray analysis (Supplementary Figure 4). In total, 6 of the 429 control subjects (1.4%) were found to be heterozygous carriers of p.R4859K. Therefore, we concluded that the p.R4859K mutation increases the risk of MMD by a remarkably high amount (odds ratio=190.8 (95% confidence interval=71.7–507.9), P=1.2 × 10−43) (Table 3). It was recently reported that an SNP (ss161110142) in the promoter region of RPTOR, which is located ∼150 kb downstream from RNF213, was associated with MMD.17 Genotyping of the SNP in RPTOR showed that the RNF213 p.R4859K mutation was more strongly associated with MMD than ss161110142 (Supplementary Figure 1).
RNF213 encodes a protein with 5256 amino acids harboring a RING (really interesting new gene) finger motif, suggesting that it functions as an E3 ubiquitin ligase (Figure 2c). It also has an AAA ATPase domain, which is characteristic of energy-dependent unfoldases.18 To our knowledge, RNF213 is the first RING finger protein known to contain an AAA ATPase domain. The expression profile of RNF213 has not been previously fully characterized. We performed a quantitative reverse transcription PCR analysis in various human tissues and cells. RNF213 mRNA was highly expressed in immune tissues, such as spleen and leukocytes (Figure 3a and Supplementary Figure 5). Expression of RNF213 was detected in fractions of both polymorphonuclear cells and mononuclear cells and was found in both B and T cell fractions (Supplementary Figure 6). A low but significant expression of RNF213 was also observed in human umbilical vein endothelial cells and human pulmonary artery smooth muscle cells. Cellular expression was not enhanced in tumor cell lines, compared with leukocytes. In human fetal tissues, the highest expression was observed in leukocytes and the thymus (Supplementary Figure 6E). The expression of RNF213 was surprisingly low in both adult and fetal brains. Overall, RNF213 was ubiquitously expressed, and the highest expression was observed in immune tissues.

Expression of human RNF213 and murine Rnf213. (a) RT-PCR analysis of RNF213 mRNA in various human tissues. The expression levels of RNF213 mRNA in various adult human tissues were evaluated by quantitative PCR using GAPDH mRNA as a control. The signal ratio of RNF213 mRNA to GAPDH mRNA in each sample is shown on the vertical axis. (b–g) In situ hybridization (ISH) analysis of Rnf213 mRNA in mouse spleen. Specific signals for Rnf213 mRNA were detected by ISH analysis with the anti-sense probe (b) but not with the sense probe (c). Hematoxylin–eosin staining of the mouse spleen (d). Signals for the Rnf213 mRNA were observed in small mononuclear cells, which were mainly localized in the white pulps (dotted square, e) and partially distributed in the red pulps (dotted squares, f and g). Panels e, f and g show the high-magnification images of the corresponding fields in panel b. Scale bars, 1 mm (b–d) and 50 μm (e–g).
We studied the cellular expression of Rnf213 in mice. The ISH analysis of spleen showed that Rnf213 mRNA was present in small mononuclear cells, which were mainly localized in the white pulps (Figures 3b–g). The ISH signals were also detected in the primary follicles in the lymph node and in thymocytes in the medulla of the thymus (Supplementary Figure 7). To study Rnf213 expression in activated lymphocytes we immunized mice with keyhole limpet hemocyanin, and examined Rnf213 mRNA in spleen by ISH analysis. Primary immunization with keyhole limpet hemocyanin antigen revealed that the expression of Rnf213 in the secondary follicle is as high as in the primary follicle in the lymph node (Supplementary Figure 8). In an E16.5 mouse embryo, expression was observed in the medulla of the thymus and in the cells around the mucous palatine glands (Supplementary Figure 9). These findings suggest that mature lymphocytes in a static state express Rnf213 mRNA at a higher level than do their immature counterparts.
Discussion
We identified a susceptibility locus for MMD by genome-wide and locus-specific association studies. Further sequencing analysis revealed a founder missense mutation in RNF213, p.R4859K, which was tightly associated with MMD onset. Identification of a founder mutation in individuals with MMD would resolve the following recurrent questions:2, 19 (i) why is MMD more prevalent in East Asia than in Western countries? The carrier frequency of p.R4859K in Japan is 1/72 (Table 2). In contrast, we found no p.R4859K carrier in 400 Caucasian controls (data not shown). Furthermore, no mutation was identified in five Caucasian patients with MMD after the full sequencing of RNF213. These results suggest that the genetic background of MMD in Asian populations is distinct from that in Western populations and that the low incidence of MMD in Western countries may be attributable to a lack of the founder RNF213 mutation. (ii) Is unilateral involvement a subtype of MMD or a different disease?2 We collected DNA samples from six patients with unilateral involvement and found a p.R4859K mutation in four of them (data not shown), suggesting that bilateral and unilateral MMD share a genetic background. (iii) Is pre-symptomatic diagnosis of MMD possible? In the present study, MMD never developed in the 15 mutation-negative family members in the 19 MMD families with the p.R4859K mutation (Table 3 and Supplementary Figure 1), suggesting the feasibility of presymptomatic diagnosis or exclusion by genetic testing.
How the mutant RNF213 protein causes MMD remains to be elucidated. The expression of RNF213 was more abundant in a subset of leukocytes than in the brain, suggesting that blood cells have a function in the etiology of MMD. This observation agrees with a previous report that MMD patients have systemic angiopathy.20 Recent studies have suggested that the postnatal vasculature can form through vasculogenesis, a process by which endothelial progenitor cell are recruited from the splenic pool and differentiate into mature endothelial cells.21 Levels of endothelial progenitor cells in the peripheral blood are increased in MMD patients.22 RNF213 may be expressed in splenic endothelial progenitor cells and mutant RNF213 might dysregulate the function of the endothelial progenitor cells. Further research is necessary to elucidate the role of RNF213 in the etiology of MMD.
References
Suzuki, J. & Takaku, A. Cerebrovascular ‘moyamoya’ disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 20, 288–299 (1969).
Suzuki, J. Moyamoya Disease (Springer-Verlag: Berlin, 1983).
Oki, K., Hoshino, H. & Suzuki, N. In: Moyamoya Disease Update, (eds Cho B.K., Tominaga T.) 29–34 (Springer: New York, 2010).
Phi, J. H., Kim, S. K., Wang, K. C. & Cho, B. K. In: Moyamoya Disease Update, (eds Cho B.K., Tominaga T). 82–86, (Springer: New York, 2010).
Yoshihara, T., Taguchi, A., Matsuyama, T., Shimizu, Y., Kikuchi-Taura, A., Soma, T. et al. Increase in circulating CD34-positive cells in patients with angiographic evidence of moyamoya-like vessels. J. Cereb. Blood Flow Metab. 28, 1086–1089 (2008).
Achrol, A. S., Guzman, R., Lee, M. & Steinberg, G. K. Pathophysiology and genetic factors in moyamoya disease. Neurosurg. Focus. 26, E4 (2009).
Scott, R. M. & Smith, E. R. Moyamoya disease and moyamoya syndrome.. N. Engl. J. Med. 360, 1226–1237 (2009).
Kure, S. In: Moyamoya Disease Update (eds Cho B.K., Tominaga T.) 41–45 (Springer: Tokyo, 2010).
Kuriyama, S., Kusaka, Y., Fujimura, M., Wakai, K., Tamakoshi, A., Hashimoto, S. et al. Prevalence and clinicoepidemiological features of moyamoya disease in Japan: findings from a nationwide epidemiological survey. Stroke. 39, 42–47 (2008).
Sakurai, K., Horiuchi, Y., Ikeda, H., Ikezaki, K., Yoshimoto, T., Fukui, M. et al. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J. Hum. Genet. 49, 278–281 (2004).
Nanba, R., Kuroda, S., Tada, M., Ishikawa, T., Houkin, K. & Iwasaki, Y. Clinical features of familial moyamoya disease. Childs. Nerv. Syst. 22, 258–262 (2006).
Ikeda, H., Sasaki, T., Yoshimoto, T., Fukui, M. & Arinami, T. Mapping of a familial moyamoya disease gene to chromosome 3p24.2-p26. Am. J. Hum. Genet. 64, 533–537 (1999).
Inoue, T. K., Ikezaki, K., Sasazuki, T., Matsushima, T. & Fukui, M. Linkage analysis of moyamoya disease on chromosome 6. J. Child. Neurol. 15, 179–182 (2000).
Yamauchi, T., Tada, M., Houkin, K., Tanaka, T., Nakamura, Y., Kuroda, S. et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25.. Stroke. 31, 930–935 (2000).
Wakai, K., Tamakoshi, A., Ikezaki, K., Fukui, M., Kawamura, T., Aoki, R. et al. Epidemiological features of moyamoya disease in Japan: findings from a nationwide survey. Clin. Neurol. Neurosurg. 99 (Suppl 2), S1–S5 (1997).
Mineharu, Y., Liu, W., Inoue, K., Matsuura, N., Inoue, S., Takenaka, K. et al. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. 70, 2357–2363 (2008).
Liu, W., Hashikata, H., Inoue, K., Matsuura, N., Mineharu, Y., Kobayashi, H. et al. A rare Asian founder polymorphism of Raptor may explain the high prevalence of Moyamoya disease among East Asians and its low prevalence among Caucasians.. Environ. Health. Prev. Med. 15, 94–104 (2010).
Lupas, A. N. & Martin, J. AAA proteins.. Curr. Opin. Struct. Biol. 12, 746–753 (2002).
Ikezaki, K. In: Moyamoya disease (eds Ikezaki K., Loftus C. M.) 43–75 (Thieme: New York, 2001).
Ikeda, E. Systemic vascular changes in spontaneous occlusion of the circle of Willis. Stroke. 22, 1358–1362 (1991).
Zampetaki, A., Kirton, J. P. & Xu, Q. Vascular repair by endothelial progenitor cells. Cardiovasc. Res. 78, 413–421 (2008).
Rafat, N., Beck, G., Pena-Tapia, P. G., Schmiedek, P. & Vajkoczy, P. Increased levels of circulating endothelial progenitor cells in patients with Moyamoya disease. Stroke. 40, 432–438 (2009).
Acknowledgements
We thank all of the patients and their families for participating in this study. We also thank Dr Hidetoshi Ikeda at the Department of Neurosurgery, Tohoku University School of Medicine and Drs Toshiaki Hayashi and Reizo Shirane at the Department of Neurosurgery, Miyagi Children's Hospital, Sendai, Japan for patient recruitment. We are grateful to Ms Kumi Kato for technical assistance. This study was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology, Japan and by the Research Committee on Moyamoya Disease of the Ministry of Health, Labor and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kamada, F., Aoki, Y., Narisawa, A. et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 56, 34–40 (2011). https://doi.org/10.1038/jhg.2010.132
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.132
This article is cited by
-
Arg4810Lys mutation in RNF213 among Eastern Indian non-MMD ischemic stroke patients: a genotype–phenotype correlation
Neurological Sciences (2024)
-
Neurovascular considerations in patients with Down syndrome and moyamoya syndrome
Child's Nervous System (2024)
-
High-resolution MRI vessel wall enhancement in moyamoya disease: risk factors and clinical outcomes
European Radiology (2024)
-
17q25.3 copy number changes: association with neurodevelopmental disorders and cardiac malformation
Molecular Cytogenetics (2023)
-
Investigation and management of pediatric moyamoya arteriopathy in the era of genotype-phenotype correlation studies
European Journal of Human Genetics (2023)
